Summary
This report analyses the Del and Het male samples only. The report begins with some general Setup and is followed by various analyses denoted by a prefix (y)
Setup
Show code
Load data
There are 25,965 features.
Cleaning up duplicate features
There are 56 features with a blank ID1. These are shown in the table below.
It’s not clear why this has happened, but such features complicate downstream analysis. To remedy this, we use the value in the Probe column to look up the corresponding Ensembl ID.
This provides an ID for 44 / 56 features.
Show code
DT::datatable(
tmp2,
caption = "Features successfully mapped to an 'ID' by the 'Probe' value.")
This leaves 12 / 56 features for which we can’t map the Probe to an ID.
Show code
DT::datatable(
tmp3,
caption = "Features that cannot be mapped to an 'ID' by the 'Probe' value.")
We opt to remove these features from the dataset.
Show code
x <- x[!is.na(x$ID), ]
Finally, there are 12 ID values that appear more than once. We pool (sum) together counts from features with the same ID.
Create DGEList objects
Show code
# NOTE: Only retain a small subset of columns since we re-annotate the
# gene-based metadata, below.
genes <- x[!duplicated(x$ID), c("ID", "Description")]
stopifnot(identical(rownames(counts), genes$ID))
y <- DGEList(counts = counts, genes = genes)
y$samples$genotype <- ifelse(grepl("het", colnames(y)), "Het", "Del")
y$samples$mouse <- substr(colnames(y), 7, 10)
y$samples$group <- y$samples$genotype
colnames(y) <- sub("\\_trim\\.bam", "", sub("Bcell\\_", "", colnames(y)))
zero_counts <- rowSums(y$counts) == 0
We exclude 7,932 genes with zero counts in all 8 samples.
Show code
y <- y[!zero_counts, ]
Incorporating gene-based annotation
We obtain gene-based annotations from the NCBI/RefSeq and Ensembl databases, such as the chromosome and gene symbol, using the Mus.musculus and EnsDb.Mmusculus.v79 packages.
Show code
# Extract rownames (Ensembl IDs) to use as key in database lookups.
ensembl <- y$genes$ID
# Pull out useful gene-based annotations from the Ensembl-based database.
library(EnsDb.Mmusculus.v79)
library(ensembldb)
# NOTE: These columns were customised for this project.
ensdb_columns <- c(
"GENEBIOTYPE", "GENENAME", "GENESEQSTART", "GENESEQEND", "SEQNAME", "SYMBOL")
names(ensdb_columns) <- paste0("ENSEMBL.", ensdb_columns)
stopifnot(all(ensdb_columns %in% columns(EnsDb.Mmusculus.v79)))
ensdb_df <- DataFrame(
lapply(ensdb_columns, function(column) {
mapIds(
x = EnsDb.Mmusculus.v79,
# NOTE: Need to remove gene version number prior to lookup.
keys = gsub("\\.[0-9]+$", "", ensembl),
keytype = "GENEID",
column = column,
multiVals = "CharacterList")
}),
row.names = ensembl)
# NOTE: Can't look up GENEID column with GENEID key, so have to add manually.
ensdb_df$ENSEMBL.GENEID <- ensembl
# NOTE: Mus.musculus combines org.Mm.eg.db and
# TxDb.Mmusculus.UCSC.mm10.knownGene (as well as others) and therefore
# uses entrez gene and RefSeq based data.
library(Mus.musculus)
# NOTE: These columns were customised for this project.
ncbi_columns <- c(
# From TxDB: None required
# From OrgDB
"ALIAS", "ENTREZID", "GENENAME", "REFSEQ", "SYMBOL")
names(ncbi_columns) <- paste0("NCBI.", ncbi_columns)
stopifnot(all(ncbi_columns %in% columns(Mus.musculus)))
ncbi_df <- DataFrame(
lapply(ncbi_columns, function(column) {
mapIds(
x = Mus.musculus,
# NOTE: Need to remove gene version number prior to lookup.
keys = gsub("\\.[0-9]+$", "", ensembl),
keytype = "ENSEMBL",
column = column,
multiVals = "CharacterList")
}),
row.names = ensembl)
y$genes <- cbind(y$genes, flattenDF(cbind(ensdb_df, ncbi_df)))
Having quantified gene expression against the Ensembl gene annotation, we have Ensembl-style identifiers for the genes. These identifiers are used as they are unambiguous and highly stable. However, they are difficult to interpret compared to the gene symbols which are more commonly used in the literature. Henceforth, we will use gene symbols (where available) to refer to genes in our analysis and otherwise use the Ensembl-style gene identifiers2.
Show code
# Replace the row names of the SCE by the gene symbols (where available).
rownames(y) <- scuttle::uniquifyFeatureNames(
ID = ensembl,
# NOTE: An Ensembl ID may map to 0, 1, 2, 3, ... gene symbols.
# When there are multiple matches only the 1st match is used.
names = sapply(strsplit(y$genes$ENSEMBL.SYMBOL, "; "), function(x) {
if (length(x)) {
x[[1]]
} else {
NA_character_
}
}))
Show code
# Some useful gene sets
mito_set <- rownames(y)[y$genes$ENSEMBL.SEQNAME == "MT"]
ribo_set <- grep("^Rp(s|l)", rownames(y), value = TRUE)
# NOTE: A more curated approach for identifying ribosomal protein genes
# (https://github.com/Bioconductor/OrchestratingSingleCellAnalysis-base/blob/ae201bf26e3e4fa82d9165d8abf4f4dc4b8e5a68/feature-selection.Rmd#L376-L380)
library(msigdbr)
c2_sets <- msigdbr(species = "Mus musculus", category = "C2")
ribo_set <- union(
ribo_set,
c2_sets[c2_sets$gs_name == "KEGG_RIBOSOME", ]$gene_symbol)
sex_set <- rownames(y)[y$genes$ENSEMBL.SEQNAME %in% c("X", "Y")]
pseudogene_set <- rownames(y)[grepl("pseudogene", y$genes$ENSEMBL.GENEBIOTYPE)]
Gene filtering
We exclude genes with insufficient counts for DE testing. Shown below are the number of genes that have sufficient counts to test for DE (TRUE) or not (FALSE).
Show code
y_keep_exprs <- filterByExpr(y, group = y$samples$group)
y <- y[y_keep_exprs, , keep.lib.sizes = FALSE]
table(y_keep_exprs)
y_keep_exprs
FALSE TRUE
4600 13409 Normalization
Normalization with upper-quartile (UQ) normalization.
Show code
y <- calcNormFactors(y, method = "upperquartile")
Analysis of male B cell samples
Show code
y_design <- model.matrix(~0 + genotype, y$samples)
colnames(y_design) <- sub("genotype", "", colnames(y_design))
MDS
Show code
plotMDS(y, col = genotype_colours[y$samples$genotype])
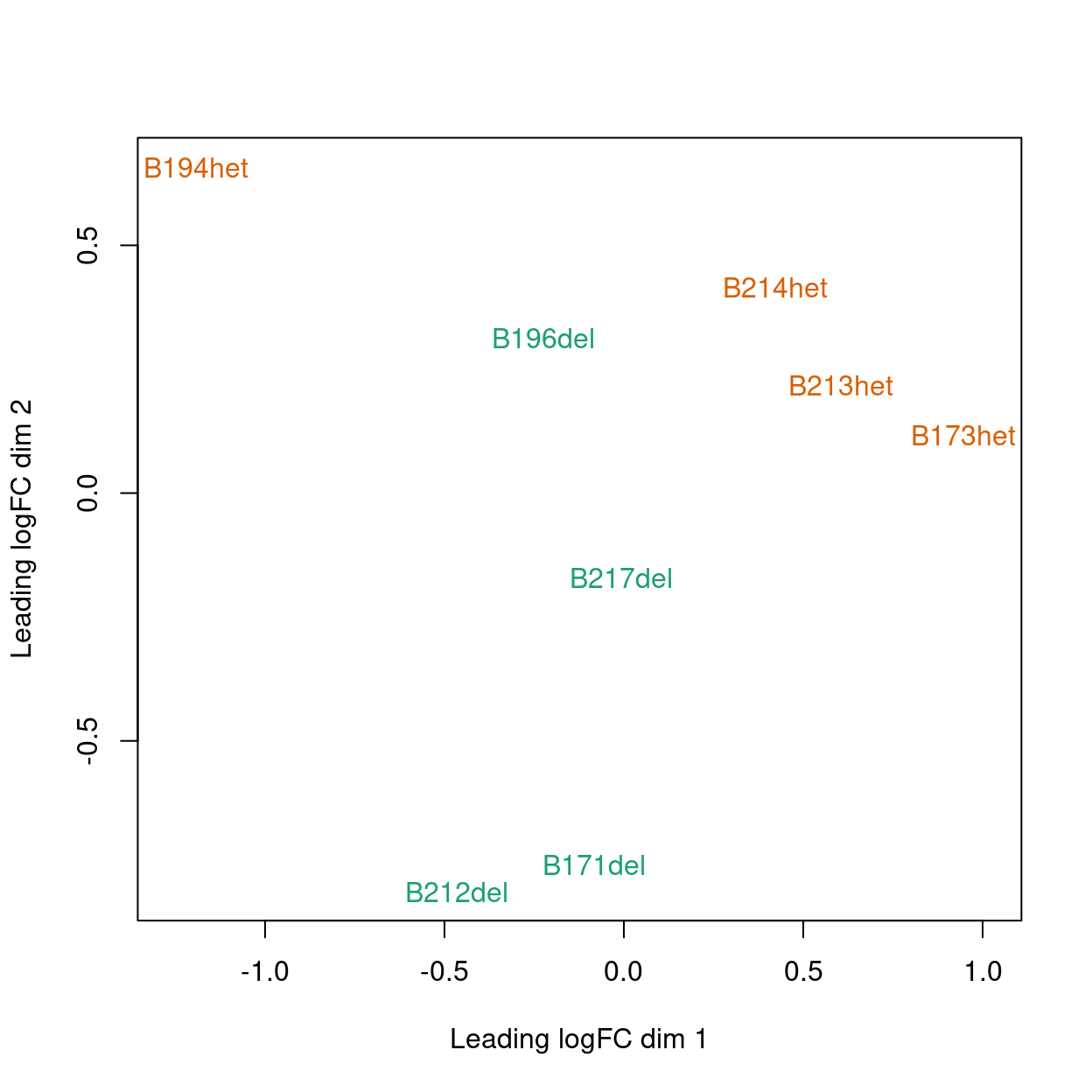
Figure 1: MDS plot coloured by genotype.
DE analysis
Fit a model with a term for genotype using the quasi-likelihood pipeline from edgeR.
Show code
y <- estimateDisp(y, y_design)
par(mfrow = c(1, 2))
plotBCV(y)
y_dgeglm <- glmQLFit(y, y_design, robust = TRUE)
plotQLDisp(y_dgeglm)
Show code
y_contrast <- makeContrasts(Del - Het, levels = y_design)
y_dgelrt <- glmQLFTest(y_dgeglm, contrast = y_contrast)
y_dt <- decideTests(y_dgelrt)
summary(y_dt) %>%
knitr::kable(caption = "Number of DEGs (FDR < 0.05)")
| 1Del -1Het | |
|---|---|
| Down | 1 |
| NotSig | 13393 |
| Up | 15 |
CSV file of results available from output/DEGs/male_B_cells/.
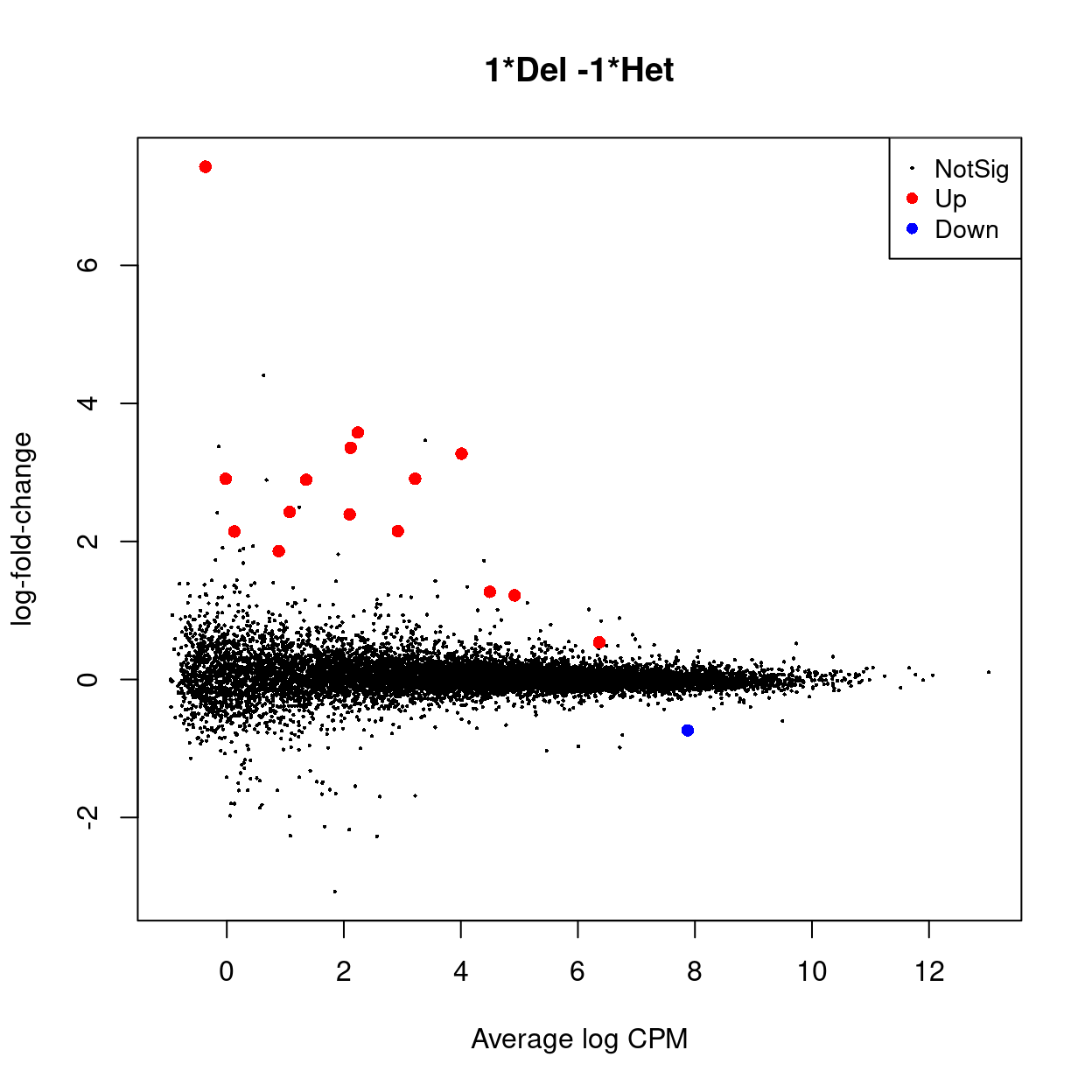
An interactive Glimma MD plot of the differential expression results is available from output/DEGs/male_B_cells/glimma-plots/male_B_cells.md-plot.html.
Show code
glMDPlot(
x = y_dgelrt,
counts = y,
anno = y$genes,
display.columns = c("ENSEMBL.SYMBOL"),
groups = y$samples$group,
samples = colnames(y),
status = y_dt,
transform = TRUE,
sample.cols = as.integer(factor(y$samples$genotype)),
path = outdir,
html = "male_B_cells.md-plot",
launch = FALSE)
Gene set tests
Gene set testing is commonly used to interpret the differential expression results in a biological context. Here we apply various gene set tests to the DEG lists.
goana
We use the goana() function from the limma R/Bioconductor package to test for over-representation of gene ontology (GO) terms in each DEG list.
Show code
list_of_go <- lapply(colnames(y_contrast), function(j) {
dgelrt <- glmLRT(y_dgeglm, contrast = y_contrast[, j])
go <- goana(
de = dgelrt,
geneid = sapply(strsplit(dgelrt$genes$NCBI.ENTREZID, "; "), "[[", 1),
species = "Mm")
gzout <- gzfile(
description = file.path(outdir, "male_B_cells.goana.csv.gz"),
open = "wb")
write.csv(
topGO(go, number = Inf),
gzout,
row.names = TRUE)
close(gzout)
go
})
names(list_of_go) <- colnames(y_contrast)
CSV files of the results are available from output/DEGs/male_B_cells/.
An example of the results are shown below.
Show code
topGO(list_of_go[[1]]) %>%
knitr::kable(
caption = '`goana()` produces a table with a row for each GO term and the following columns: **Term** (GO term); **Ont** (ontology that the GO term belongs to. Possible values are "BP", "CC" and "MF"); **N** (number of genes in the GO term); **Up** (number of up-regulated differentially expressed genes); **Down** (number of down-regulated differentially expressed genes); **P.Up** (p-value for over-representation of GO term in up-regulated genes); **P.Down** (p-value for over-representation of GO term in down-regulated genes)')
| Term | Ont | N | Up | Down | P.Up | P.Down | |
|---|---|---|---|---|---|---|---|
| GO:0031720 | haptoglobin binding | MF | 4 | 0 | 4 | 1.0000000 | 0.0000000 |
| GO:0005833 | hemoglobin complex | CC | 4 | 0 | 4 | 1.0000000 | 0.0000000 |
| GO:0048731 | system development | BP | 2947 | 31 | 31 | 0.0131091 | 0.0000000 |
| GO:0030426 | growth cone | CC | 164 | 1 | 10 | 0.6976242 | 0.0000000 |
| GO:0030427 | site of polarized growth | CC | 170 | 1 | 10 | 0.7106582 | 0.0000000 |
| GO:0031838 | haptoglobin-hemoglobin complex | CC | 5 | 1 | 4 | 0.0355835 | 0.0000000 |
| GO:0007275 | multicellular organism development | BP | 3301 | 34 | 31 | 0.0121146 | 0.0000000 |
| GO:0048821 | erythrocyte development | BP | 38 | 0 | 6 | 1.0000000 | 0.0000000 |
| GO:0048468 | cell development | BP | 1486 | 16 | 21 | 0.0652232 | 0.0000000 |
| GO:0007399 | nervous system development | BP | 1490 | 17 | 21 | 0.0364729 | 0.0000000 |
| GO:0019825 | oxygen binding | MF | 8 | 0 | 4 | 1.0000000 | 0.0000000 |
| GO:0150034 | distal axon | CC | 247 | 2 | 10 | 0.5353054 | 0.0000000 |
| GO:0044297 | cell body | CC | 522 | 3 | 13 | 0.7331790 | 0.0000000 |
| GO:0048856 | anatomical structure development | BP | 3597 | 36 | 31 | 0.0148414 | 0.0000000 |
| GO:0032502 | developmental process | BP | 3893 | 36 | 32 | 0.0477809 | 0.0000000 |
| GO:0005344 | oxygen carrier activity | MF | 3 | 0 | 3 | 1.0000000 | 0.0000000 |
| GO:0048858 | cell projection morphogenesis | BP | 453 | 8 | 12 | 0.0164555 | 0.0000000 |
| GO:0032501 | multicellular organismal process | BP | 4220 | 42 | 33 | 0.0077364 | 0.0000001 |
| GO:0001944 | vasculature development | BP | 494 | 17 | 3 | 0.0000001 | 0.2635084 |
| GO:0032990 | cell part morphogenesis | BP | 474 | 8 | 12 | 0.0209935 | 0.0000001 |
kegga
We use the kegga() function from the limma R/Bioconductor package to test for over-representation of KEGG pathways in each DEG list.
Show code
list_of_kegg <- lapply(colnames(y_contrast), function(j) {
dgelrt <- glmLRT(y_dgeglm, contrast = y_contrast[, j])
kegg <- kegga(
de = dgelrt,
geneid = sapply(strsplit(dgelrt$genes$NCBI.ENTREZID, "; "), "[[", 1),
species = "Mm")
gzout <- gzfile(
description = file.path(outdir, "male_B_cells.kegga.csv.gz"),
open = "wb")
write.csv(
topKEGG(kegg, number = Inf),
gzout,
row.names = TRUE)
close(gzout)
kegg
})
names(list_of_kegg) <- colnames(y_contrast)
CSV files of the results are available from output/DEGs/male_B_cells/.
An example of the results are shown below.
Show code
topKEGG(list_of_kegg[[1]]) %>%
knitr::kable(
caption = '`kegga()` produces a table with a row for each KEGG pathway ID and the following columns: **Pathway** (KEGG pathway); **N** (number of genes in the GO term); **Up** (number of up-regulated differentially expressed genes); **Down** (number of down-regulated differentially expressed genes); **P.Up** (p-value for over-representation of KEGG pathway in up-regulated genes); **P.Down** (p-value for over-representation of KEGG pathway in down-regulated genes)')
| Pathway | N | Up | Down | P.Up | P.Down | |
|---|---|---|---|---|---|---|
| path:mmu05143 | African trypanosomiasis | 26 | 1 | 4 | 0.1735812 | 0.0000021 |
| path:mmu04668 | TNF signaling pathway | 92 | 7 | 0 | 0.0000046 | 1.0000000 |
| path:mmu04380 | Osteoclast differentiation | 111 | 7 | 0 | 0.0000160 | 1.0000000 |
| path:mmu05144 | Malaria | 46 | 1 | 4 | 0.2865069 | 0.0000216 |
| path:mmu04657 | IL-17 signaling pathway | 57 | 5 | 0 | 0.0000576 | 1.0000000 |
| path:mmu05418 | Fluid shear stress and atherosclerosis | 112 | 6 | 0 | 0.0001647 | 1.0000000 |
| path:mmu01522 | Endocrine resistance | 78 | 5 | 0 | 0.0002573 | 1.0000000 |
| path:mmu04915 | Estrogen signaling pathway | 79 | 5 | 1 | 0.0002731 | 0.2491464 |
| path:mmu04935 | Growth hormone synthesis, secretion and action | 88 | 5 | 0 | 0.0004509 | 1.0000000 |
| path:mmu05133 | Pertussis | 54 | 4 | 0 | 0.0006355 | 1.0000000 |
| path:mmu04713 | Circadian entrainment | 57 | 4 | 0 | 0.0007807 | 1.0000000 |
| path:mmu05224 | Breast cancer | 101 | 5 | 0 | 0.0008464 | 1.0000000 |
| path:mmu05030 | Cocaine addiction | 30 | 3 | 0 | 0.0013240 | 1.0000000 |
| path:mmu05200 | Pathways in cancer | 376 | 9 | 2 | 0.0016617 | 0.3953707 |
| path:mmu04024 | cAMP signaling pathway | 118 | 5 | 0 | 0.0016969 | 1.0000000 |
| path:mmu05135 | Yersinia infection | 123 | 5 | 0 | 0.0020368 | 1.0000000 |
| path:mmu04620 | Toll-like receptor signaling pathway | 78 | 4 | 0 | 0.0025097 | 1.0000000 |
| path:mmu05142 | Chagas disease | 83 | 4 | 0 | 0.0031451 | 1.0000000 |
| path:mmu04670 | Leukocyte transendothelial migration | 83 | 4 | 0 | 0.0031451 | 1.0000000 |
| path:mmu04926 | Relaxin signaling pathway | 86 | 4 | 0 | 0.0035748 | 1.0000000 |
camera with MSigDB gene sets
We use the camera() function from the limma R/Bioconductor package to test whether a set of genes is highly ranked relative to other genes in terms of differential expression, accounting for inter-gene correlation. Specifically, we test using gene sets from MSigDB, namely:
- H: hallmark gene sets are coherently expressed signatures derived by aggregating many MSigDB gene sets to represent well-defined biological states or processes.
- C2: curated gene sets from online pathway databases, publications in PubMed, and knowledge of domain experts.
Show code
# NOTE: Using BiocFileCache to avoid re-downloading these gene sets everytime
# the report is rendered.
library(BiocFileCache)
bfc <- BiocFileCache()
# NOTE: Creating list of gene sets in this slightly convoluted way so that each
# gene set name is prepended by its origin (e.g. H, C2, or C7).
msigdb <- do.call(
c,
list(
H = readRDS(
bfcrpath(
bfc,
"http://bioinf.wehi.edu.au/MSigDB/v7.1/Mm.h.all.v7.1.entrez.rds")),
C2 = readRDS(
bfcrpath(
bfc,
"http://bioinf.wehi.edu.au/MSigDB/v7.1/Mm.c2.all.v7.1.entrez.rds"))))
y_idx <- ids2indices(
msigdb,
id = sapply(strsplit(y$genes$NCBI.ENTREZID, "; "), "[[", 1))
list_of_camera <- lapply(colnames(y_contrast), function(j) {
cam <- camera(
y = y,
index = y_idx,
design = y_design,
contrast = y_contrast[, j])
gzout <- gzfile(
description = file.path(outdir, "male_B_cells.camera.csv.gz"),
open = "wb")
write.csv(
cam,
gzout,
row.names = TRUE)
close(gzout)
cam
})
names(list_of_camera) <- colnames(y_contrast)
CSV files of the results are available from output/DEGs/male_B_cells.
An example of the results are shown below.
Show code
head(list_of_camera[[1]]) %>%
knitr::kable(
caption = '`camera()` produces a table with a row for each gene set (prepended by which MSigDB collection it comes from) and the following columns: **NGenes** (number of genes in set); **Direction** (direction of change); **PValue** (two-tailed p-value); **FDR** (Benjamini and Hochberg FDR adjusted p-value).')
| NGenes | Direction | PValue | FDR | |
|---|---|---|---|---|
| C2.ZHOU_PANCREATIC_EXOCRINE_PROGENITOR | 3 | Up | 0 | 0e+00 |
| C2.MORI_MATURE_B_LYMPHOCYTE_DN | 92 | Down | 0 | 0e+00 |
| C2.REACTOME_EUKARYOTIC_TRANSLATION_ELONGATION | 108 | Up | 0 | 0e+00 |
| C2.KEGG_RIBOSOME | 103 | Up | 0 | 0e+00 |
| C2.LEE_EARLY_T_LYMPHOCYTE_UP | 116 | Down | 0 | 1e-07 |
| C2.REACTOME_RESPONSE_OF_EIF2AK4_GCN2_TO_AMINO_ACID_DEFICIENCY | 117 | Up | 0 | 2e-07 |
Sarah’s gene lists
Show code
y_X <- y$genes$ENSEMBL.SEQNAME == "X"
y_index <- list(`X-linked` = y_X)
Sarah requested testing all X-linked genes as a gene set.
We use the fry() function from the edgeR R/Bioconductor package to perform a self-contained gene set test against the null hypothesis that none of the genes in the set are differentially expressed.
Show code
fry(
y,
index = y_index,
design = y_design,
contrast = y_contrast,
sort = "none") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| X-linked | 416 | Down | 0.9861314 | 0.6496793 |
Show code
geneSetTestFiguresForPaper <- function() {
for (n in names(y_index)) {
if (is.data.frame(y_index[[n]])) {
y_status <- rep(0, nrow(y_dgelrt))
y_status[rownames(y) %in% y_index[[n]][, 1]] <-
sign(y_index[[n]][y_index[[n]][, 1] %in% rownames(y), 2])
plotMD(
y_dgelrt,
status = y_status,
hl.col = c("red", "blue"),
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
} else {
y_status <- rep("Other", nrow(y_dgelrt))
y_status[y_index[[n]]] <- n
plotMD(
y_dgelrt,
status = y_status,
values = n,
hl.col = "red",
legend = "topright",
main = n)
abline(h = 0, col = "darkgrey")
}
if (is.data.frame(y_index[[n]])) {
barcodeplot(
y_dgelrt$table$logFC,
rownames(y) %in% y_index[[n]][, 1],
gene.weights = y_index[[n]][
y_index[[n]][, 1] %in% rownames(y), 2])
} else {
barcodeplot(
y_dgelrt$table$logFC,
y_index[[n]])
}
}
}
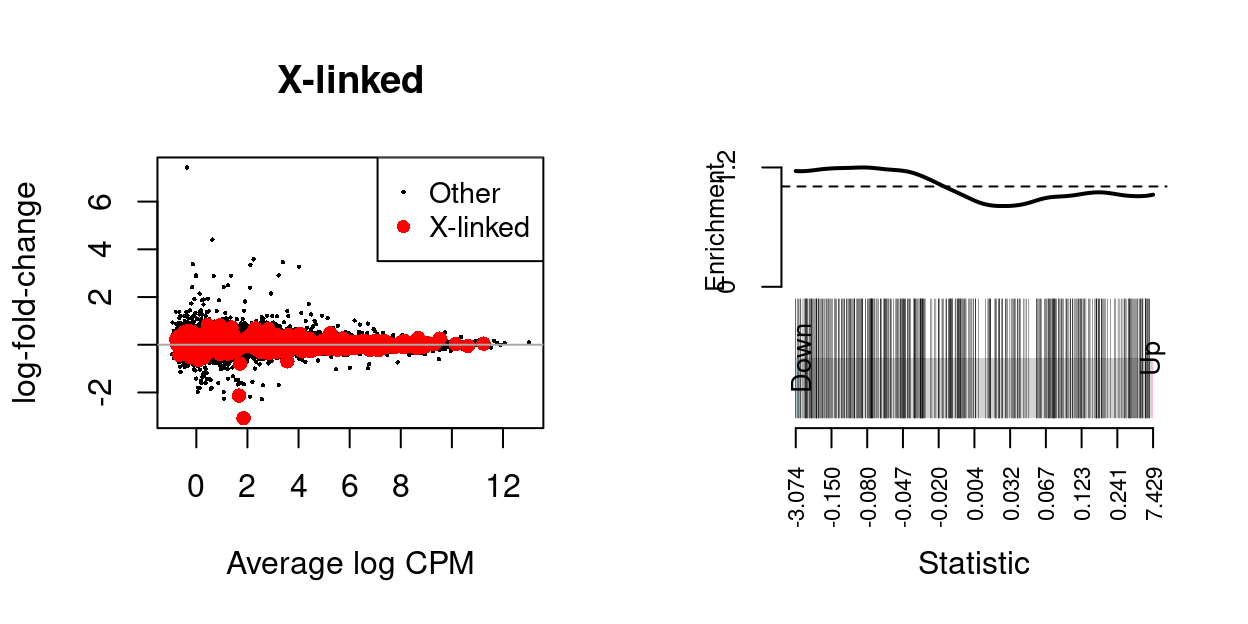
Figure 3: MD-plot and barcode plot of genes in supplied gene sets. Directional gene sets have (1) MD plot points coloured according to the statistical significance of the gene in the gene set (2) barcode plot ‘weights’ given by the logFC of the gene in the gene set. For the barcode plot, genes are represented by bars and are ranked from left to right by increasing log-fold change. This forms the barcode-like pattern. The line (or worm) above the barcode shows the relative local enrichment of the vertical bars in each part of the plot. The dotted horizontal line indicates neutral enrichment; the worm above the dotted line shows enrichment while the worm below the dotted line shows depletion.
Session info
Show code
sessioninfo::session_info()
─ Session info ─────────────────────────────────────────────────────
setting value
version R version 4.0.3 (2020-10-10)
os CentOS Linux 7 (Core)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_US.UTF-8
ctype en_US.UTF-8
tz Australia/Melbourne
date 2021-10-20
─ Packages ─────────────────────────────────────────────────────────
! package * version date lib
P annotate 1.68.0 2020-10-27 [?]
P AnnotationDbi * 1.52.0 2020-10-27 [?]
P AnnotationFilter * 1.14.0 2020-10-27 [?]
P askpass 1.1 2019-01-13 [?]
P assertthat 0.2.1 2019-03-21 [?]
P beachmat 2.6.4 2020-12-20 [?]
P Biobase * 2.50.0 2020-10-27 [?]
P BiocFileCache * 1.14.0 2020-10-27 [?]
P BiocGenerics * 0.36.0 2020-10-27 [?]
P BiocManager 1.30.10 2019-11-16 [?]
P BiocParallel 1.24.1 2020-11-06 [?]
P BiocStyle 2.18.1 2020-11-24 [?]
P biomaRt 2.46.3 2021-02-09 [?]
P Biostrings 2.58.0 2020-10-27 [?]
P bit 4.0.4 2020-08-04 [?]
P bit64 4.0.5 2020-08-30 [?]
P bitops 1.0-6 2013-08-17 [?]
P blob 1.2.1 2020-01-20 [?]
P cachem 1.0.4 2021-02-13 [?]
P cli 2.3.1 2021-02-23 [?]
P colorspace 2.0-0 2020-11-11 [?]
P crayon 1.4.1 2021-02-08 [?]
P crosstalk 1.1.1 2021-01-12 [?]
P curl 4.3 2019-12-02 [?]
P data.table 1.14.0 2021-02-21 [?]
P DBI 1.1.1 2021-01-15 [?]
P dbplyr * 2.1.0 2021-02-03 [?]
P DelayedArray 0.16.1 2021-01-22 [?]
P DelayedMatrixStats 1.12.3 2021-02-03 [?]
P DESeq2 1.30.1 2021-02-19 [?]
P digest 0.6.27 2020-10-24 [?]
P distill 1.2 2021-01-13 [?]
P downlit 0.2.1 2020-11-04 [?]
P dplyr 1.0.4 2021-02-02 [?]
P DT 0.17 2021-01-06 [?]
P edgeR * 3.32.1 2021-01-14 [?]
P ellipsis 0.3.1 2020-05-15 [?]
P EnsDb.Mmusculus.v79 * 2.99.0 2020-07-15 [?]
P ensembldb * 2.14.0 2020-10-27 [?]
P evaluate 0.14 2019-05-28 [?]
P fansi 0.4.2 2021-01-15 [?]
P fastmap 1.1.0 2021-01-25 [?]
P genefilter 1.72.1 2021-01-21 [?]
P geneplotter 1.68.0 2020-10-27 [?]
P generics 0.1.0 2020-10-31 [?]
P GenomeInfoDb * 1.26.2 2020-12-08 [?]
P GenomeInfoDbData 1.2.4 2020-10-20 [?]
P GenomicAlignments 1.26.0 2020-10-27 [?]
P GenomicFeatures * 1.42.1 2020-11-12 [?]
P GenomicRanges * 1.42.0 2020-10-27 [?]
P ggplot2 3.3.3 2020-12-30 [?]
P Glimma * 2.0.0 2020-10-27 [?]
P glue 1.4.2 2020-08-27 [?]
P GO.db * 3.12.1 2020-11-04 [?]
P graph 1.68.0 2020-10-27 [?]
P gtable 0.3.0 2019-03-25 [?]
P here * 1.0.1 2020-12-13 [?]
P highr 0.8 2019-03-20 [?]
P hms 1.0.0 2021-01-13 [?]
P htmltools 0.5.1.1 2021-01-22 [?]
P htmlwidgets 1.5.3 2020-12-10 [?]
P httr 1.4.2 2020-07-20 [?]
P IRanges * 2.24.1 2020-12-12 [?]
P jsonlite 1.7.2 2020-12-09 [?]
P knitr 1.31 2021-01-27 [?]
P lattice 0.20-41 2020-04-02 [3]
P lazyeval 0.2.2 2019-03-15 [?]
P lifecycle 1.0.0 2021-02-15 [?]
P limma * 3.46.0 2020-10-27 [?]
P locfit 1.5-9.4 2020-03-25 [?]
P magrittr 2.0.1 2020-11-17 [?]
P Matrix 1.2-18 2019-11-27 [3]
P MatrixGenerics 1.2.1 2021-01-30 [?]
P matrixStats 0.58.0 2021-01-29 [?]
P memoise 2.0.0 2021-01-26 [?]
P msigdbr * 7.2.1 2020-10-02 [?]
P munsell 0.5.0 2018-06-12 [?]
P Mus.musculus * 1.3.1 2020-07-15 [?]
P openssl 1.4.3 2020-09-18 [?]
P org.Mm.eg.db * 3.12.0 2020-10-20 [?]
P OrganismDbi * 1.32.0 2020-10-27 [?]
P pillar 1.5.0 2021-02-22 [?]
P pkgconfig 2.0.3 2019-09-22 [?]
P prettyunits 1.1.1 2020-01-24 [?]
P progress 1.2.2 2019-05-16 [?]
P ProtGenerics 1.22.0 2020-10-27 [?]
P purrr 0.3.4 2020-04-17 [?]
P R6 2.5.0 2020-10-28 [?]
P rappdirs 0.3.3 2021-01-31 [?]
P RBGL 1.66.0 2020-10-27 [?]
P RColorBrewer 1.1-2 2014-12-07 [?]
P Rcpp 1.0.6 2021-01-15 [?]
P RCurl 1.98-1.2 2020-04-18 [?]
P rlang 0.4.10 2020-12-30 [?]
P rmarkdown 2.11 2021-09-14 [?]
P rprojroot 2.0.2 2020-11-15 [?]
P Rsamtools 2.6.0 2020-10-27 [?]
P RSQLite 2.2.3 2021-01-24 [?]
P rtracklayer 1.50.0 2020-10-27 [?]
P S4Vectors * 0.28.1 2020-12-09 [?]
P scales * 1.1.1 2020-05-11 [?]
P scuttle 1.0.4 2020-12-17 [?]
P sessioninfo 1.1.1 2018-11-05 [?]
P SingleCellExperiment 1.12.0 2020-10-27 [?]
P sparseMatrixStats 1.2.1 2021-02-02 [?]
P statmod 1.4.35 2020-10-19 [?]
P stringi 1.5.3 2020-09-09 [?]
P stringr 1.4.0 2019-02-10 [?]
P SummarizedExperiment 1.20.0 2020-10-27 [?]
P survival 3.2-7 2020-09-28 [3]
P tibble 3.0.6 2021-01-29 [?]
P tidyselect 1.1.0 2020-05-11 [?]
P TxDb.Mmusculus.UCSC.mm10.knownGene * 3.10.0 2020-07-15 [?]
P utf8 1.1.4 2018-05-24 [?]
P vctrs 0.3.6 2020-12-17 [?]
P withr 2.4.1 2021-01-26 [?]
P xfun 0.26 2021-09-14 [?]
P XML 3.99-0.5 2020-07-23 [?]
P xml2 1.3.2 2020-04-23 [?]
P xtable 1.8-4 2019-04-21 [?]
P XVector 0.30.0 2020-10-27 [?]
P yaml 2.2.1 2020-02-01 [?]
P zlibbioc 1.36.0 2020-10-27 [?]
source
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.0)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.2)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.2)
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
Bioconductor
Bioconductor
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
Bioconductor
Bioconductor
CRAN (R 4.0.2)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
Bioconductor
[1] /stornext/Projects/score/Analyses/C086_Kinkel/renv/library/R-4.0/x86_64-pc-linux-gnu
[2] /tmp/RtmpfgojZ6/renv-system-library
[3] /stornext/System/data/apps/R/R-4.0.3/lib64/R/library
P ── Loaded and on-disk path mismatch.