Show code
library(SingleCellExperiment)
library(here)
library(rmarkdown)
library(ggplot2)
library(patchwork)
library(BiocParallel)
library(scater)
library(cowplot)
library(limma)
library(scales)
# NOTE: Using >= 4 cores seizes up my laptop. Can use more on RStudio server.
options(
"mc.cores" =
ifelse(Sys.info()[["nodename"]] == "rstudio-1.hpc.wehi.edu.au", 8L, 2L))
register(MulticoreParam(workers = getOption("mc.cores")))
knitr::opts_chunk$set(fig.path = "C086_Kinkel.preprocess_files/")
Setting up the data
We start from the count matrices created in code/scPipe.R, specifically the UMI counts (a.k.a. UMI-deduplicated) data and read counts (a.k.a. not UMI-deduplicated) data.
Show code
sce_deduped <- readRDS(
here("data", "SCEs", "C086_Kinkel.UMI_deduped.scPipe.SCE.rds"))
sce_not_deduped <- readRDS(
here("data", "SCEs", "C086_Kinkel.not_UMI_deduped.scPipe.SCE.rds"))
colnames(sce_not_deduped) <- sub(
"\\.not_UMI_deduped",
"",
colnames(sce_not_deduped))
altExp(sce_not_deduped, "ERCC") <- altExp(
sce_not_deduped,
"ERCC")[rownames(altExp(sce_deduped, "ERCC"))]
stopifnot(
identical(rownames(sce_deduped), rownames(sce_not_deduped)),
identical(colnames(sce_deduped), colnames(sce_not_deduped)),
identical(
colnames(altExp(sce_deduped, "ERCC")),
colnames(altExp(sce_not_deduped, "ERCC"))),
identical(
rownames(altExp(sce_deduped, "ERCC")),
rownames(altExp(sce_not_deduped, "ERCC"))))
# Combine UMI and read counts a single SCE.
sce <- sce_deduped
assay(sce, "UMI_counts") <- assay(sce_deduped, "counts")
assay(sce, "read_counts") <- assay(sce_not_deduped, "counts")
assay(altExp(sce, "ERCC"), "UMI_counts") <- assay(
altExp(sce_deduped, "ERCC"),
"counts")
assay(altExp(sce, "ERCC"), "read_counts") <- assay(
altExp(sce_not_deduped, "ERCC"),
"counts")
# NOTE: Nullify some now-unrequired data.
assay(sce, "counts") <- NULL
assay(altExp(sce, "ERCC"), "counts") <- NULL
sce$UMI_deduped <- NULL
We combine these into a single object1 containing both the UMI counts and the read counts for 25587 genes and 36 samples.
Read mapping metrics
Motivation
We aimed to sequence 5 million reads/sample and hoped that the majority of these reads would be exonic.
Analysis
Show code
library(scPipe)
library(tidyr)
library(dplyr)
qc_df <- as.data.frame(QC_metrics(sce_deduped)) %>%
tibble::rownames_to_column(var = "rowname") %>%
pivot_longer(!rowname) %>%
mutate(
well_position = sub("LCE[0-9]+_", "", rowname),
plate_number = substr(rowname, 1, 6)) %>%
inner_join(
as.data.frame(colData(sce_deduped)) %>%
tibble::rownames_to_column(var = "rowname")) %>%
mutate(well_position = factor(
well_position,
unlist(
lapply(LETTERS[1:16], function(x) paste0(x, 1:24)),
use.names = TRUE)))
Figure 1 shows that we generated more than 5 million reads for the majority of samples and that most of these reads are exonic. As expected, the negative control sample generated very few reads.
Show code
mapped_category_colours <- setNames(
RColorBrewer::brewer.pal(
length(unique(qc_df$name)),
"Dark2"),
sort(unique(qc_df$name)))
ggplot(qc_df) +
geom_col(aes(x = well_position, fill = name, y = value)) +
cowplot::theme_cowplot(font_size = 8) +
ylab("Number of reads") +
xlab("Well position") +
geom_hline(yintercept = 5e6, lty = 2) +
scale_fill_manual(values = mapped_category_colours) +
facet_grid(sample_name ~ .) +
ggtitle(
"C086_Kinkel: Read mapping metrics",
subtitle = "Number of reads per category") +
scale_x_discrete(guide = guide_axis(n.dodge = 2)) +
theme(axis.text.x = element_text(size = 6)) +
guides(fill = guide_legend(title = "Mapping category"))
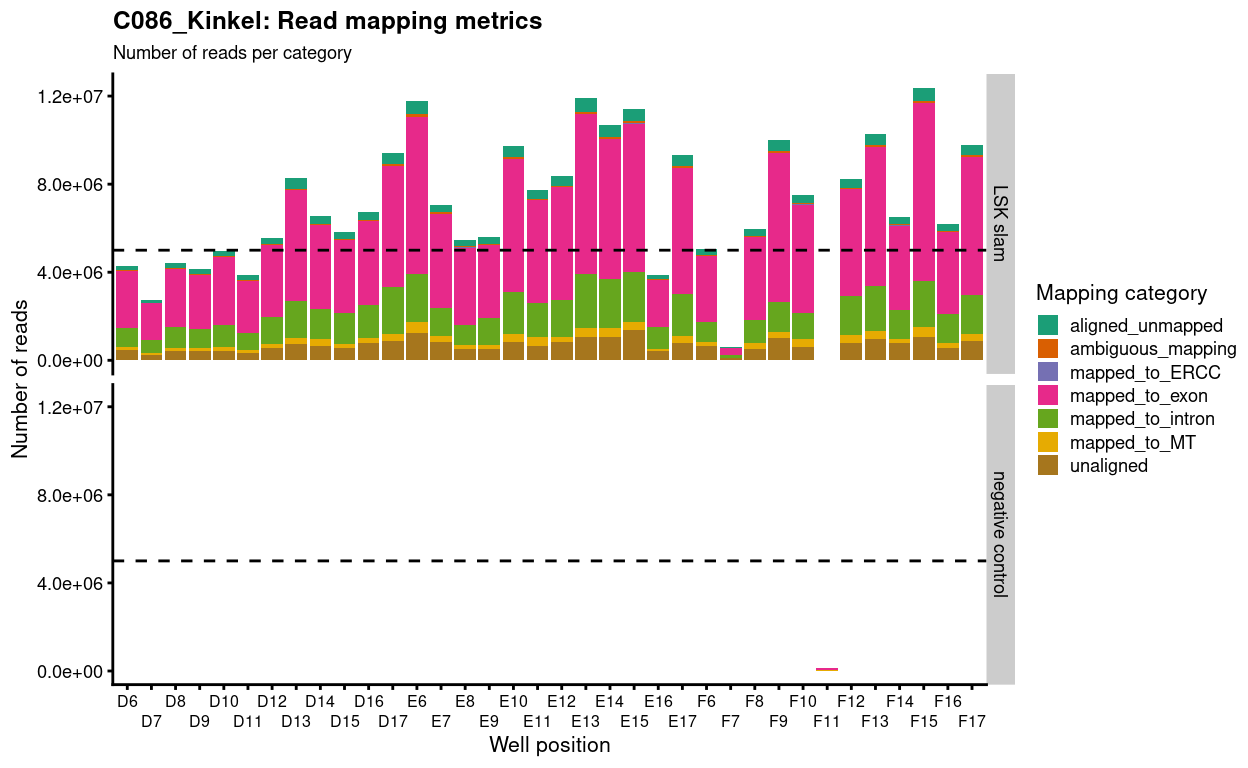
Figure 1: Number of reads generated per sample. The dashed horizional line denotes 5 million reads.
Figure 2 shows for each sample that around 60% of reads are exonic and a further 20% are intronic.
Show code
ggplot(qc_df) +
geom_col(aes(x = well_position, fill = name, y = value), position = "fill") +
cowplot::theme_cowplot(font_size = 8) +
ylab("Proportion of reads") +
xlab("Well position") +
scale_fill_manual(values = mapped_category_colours) +
facet_grid(plate_number + sample_name ~ .) +
ggtitle(
"C086_Kinkel: Read mapping metrics",
subtitle = "Proportion of reads per category") +
scale_x_discrete(guide = guide_axis(n.dodge = 2)) +
theme(axis.text.x = element_text(size = 6)) +
guides(fill = guide_legend(title = "Mapping category"))
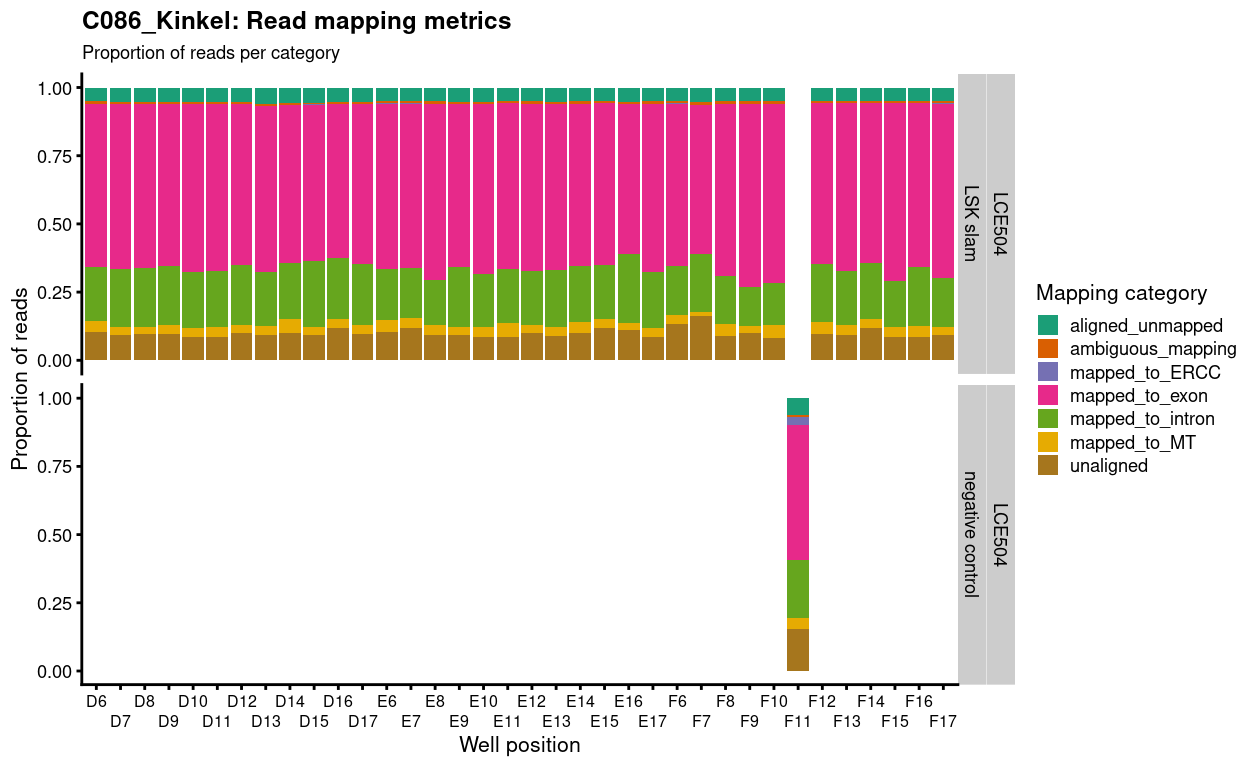
Figure 2: Proportion of reads generated in each mapping category per sample.
Figure 3 shows for each sample that on average 4 million reads are exonic and a further 1 million are intronic
Show code
qc_df %>%
ggplot() +
geom_boxplot(aes(x = name, y = value, fill = name)) +
cowplot::theme_cowplot(font_size = 12) +
ylab("Number of reads") +
xlab("Mapping category") +
scale_fill_manual(values = mapped_category_colours) +
ggtitle(
"C086_Kinkel: Number of reads per category",
subtitle = "Includes negative control sample") +
guides(fill = FALSE) +
scale_x_discrete(guide = guide_axis(angle = 45))
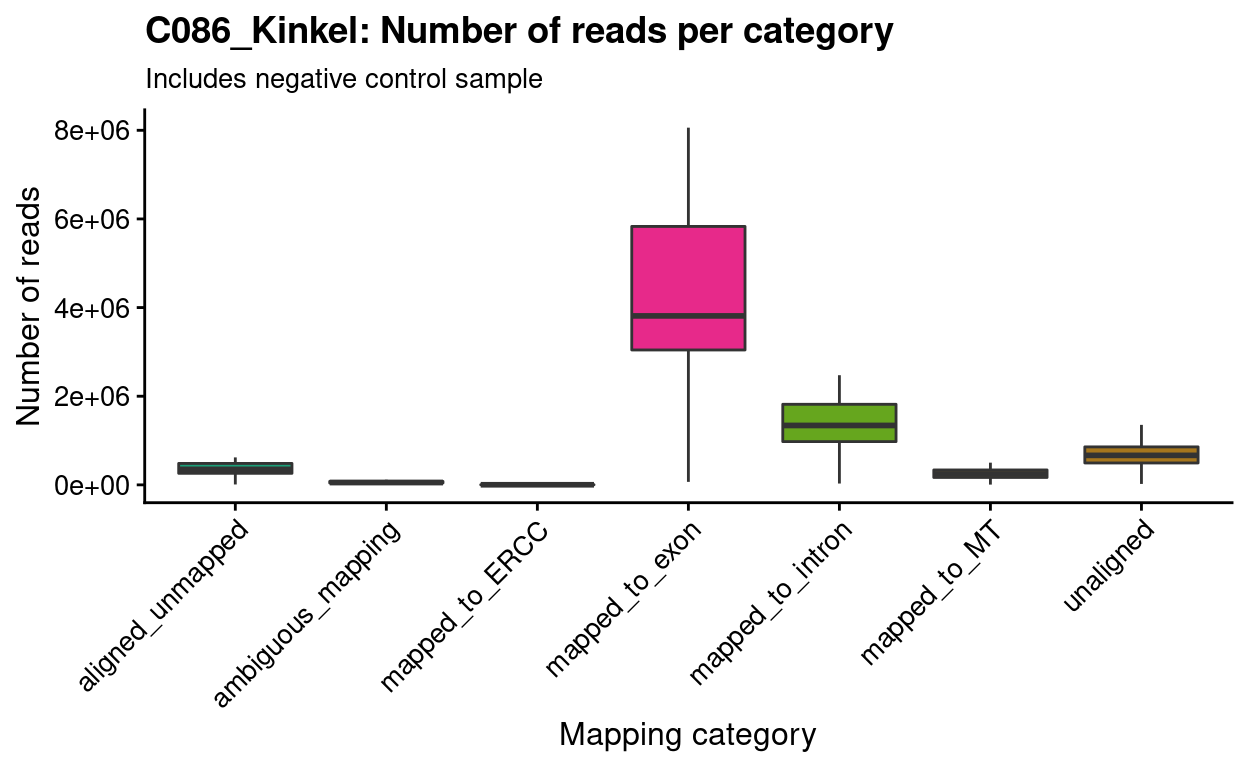
Figure 3: Number of exonic and intronic reads generated per sample.
Summary
We largely achieved our aim of sequencing ~5 million reads/sample and with the majority of reads being exonic. Henceforth, we only use the exonic reads. Although the intronic reads are potentially useful, to do so would require substantial changes to the data analysis workflow and additional analysis.
We now remove the negative control sample.
Show code
sce <- sce[, sce$sample_name != "negative control"]
colData(sce) <- droplevels(colData(sce))
We look more closely at which samples have smaller numbers of exonic reads in Have we sequenced enough? and Quality control of samples.
Incorporating metadata
Incorporating gene-based annotation
We obtain gene-based annotations from the NCBI/RefSeq and Ensembl databases, such as the chromosome and gene symbol, using the Mus.musculus and EnsDb.Mmusculus.v79 packages.
Show code
# Extract rownames (Ensembl IDs) to use as key in database lookups.
ensembl <- rownames(sce)
# Pull out useful gene-based annotations from the Ensembl-based database.
library(EnsDb.Mmusculus.v79)
library(ensembldb)
# NOTE: These columns were customised for this project.
ensdb_columns <- c(
"GENEBIOTYPE", "GENENAME", "GENESEQSTART", "GENESEQEND", "SEQNAME", "SYMBOL")
names(ensdb_columns) <- paste0("ENSEMBL.", ensdb_columns)
stopifnot(all(ensdb_columns %in% columns(EnsDb.Mmusculus.v79)))
ensdb_df <- DataFrame(
lapply(ensdb_columns, function(column) {
mapIds(
x = EnsDb.Mmusculus.v79,
# NOTE: Need to remove gene version number prior to lookup.
keys = gsub("\\.[0-9]+$", "", ensembl),
keytype = "GENEID",
column = column,
multiVals = "CharacterList")
}),
row.names = ensembl)
# NOTE: Can't look up GENEID column with GENEID key, so have to add manually.
ensdb_df$ENSEMBL.GENEID <- ensembl
# NOTE: Mus.musculus combines org.Mm.eg.db and
# TxDb.Mmusculus.UCSC.mm10.knownGene (as well as others) and therefore
# uses entrez gene and RefSeq based data.
library(Mus.musculus)
# NOTE: These columns were customised for this project.
ncbi_columns <- c(
# From TxDB: None required
# From OrgDB
"ALIAS", "ENTREZID", "GENENAME", "REFSEQ", "SYMBOL")
names(ncbi_columns) <- paste0("NCBI.", ncbi_columns)
stopifnot(all(ncbi_columns %in% columns(Mus.musculus)))
ncbi_df <- DataFrame(
lapply(ncbi_columns, function(column) {
mapIds(
x = Mus.musculus,
# NOTE: Need to remove gene version number prior to lookup.
keys = gsub("\\.[0-9]+$", "", ensembl),
keytype = "ENSEMBL",
column = column,
multiVals = "CharacterList")
}),
row.names = ensembl)
rowData(sce) <- cbind(ensdb_df, ncbi_df)
Having quantified gene expression against the GENCODE gene annotation, we have Ensembl-style identifiers for the genes. These identifiers are used as they are unambiguous and highly stable. However, they are difficult to interpret compared to the gene symbols which are more commonly used in the literature. Henceforth, we will use gene symbols (where available) to refer to genes in our analysis and otherwise use the Ensembl-style gene identifiers2.
Show code
# Replace the row names of the SCE by the gene symbols (where available).
rownames(sce) <- uniquifyFeatureNames(
ID = rownames(sce),
# NOTE: An Ensembl ID may map to 0, 1, 2, 3, ... gene symbols.
# When there are multiple matches only the 1st match is used.
names = sapply(rowData(sce)$ENSEMBL.SYMBOL, function(x) {
if (length(x)) {
x[[1]]
} else {
NA_character_
}
}))
Show code
# Some useful gene sets
mito_set <- rownames(sce)[any(rowData(sce)$ENSEMBL.SEQNAME == "MT")]
ribo_set <- grep("^Rp(s|l)", rownames(sce), value = TRUE)
# NOTE: A more curated approach for identifying ribosomal protein genes
# (https://github.com/Bioconductor/OrchestratingSingleCellAnalysis-base/blob/ae201bf26e3e4fa82d9165d8abf4f4dc4b8e5a68/feature-selection.Rmd#L376-L380)
library(msigdbr)
c2_sets <- msigdbr(species = "Mus musculus", category = "C2")
ribo_set <- union(
ribo_set,
c2_sets[c2_sets$gs_name == "KEGG_RIBOSOME", ]$gene_symbol)
sex_set <- rownames(sce)[any(rowData(sce)$ENSEMBL.SEQNAME %in% c("X", "Y"))]
pseudogene_set <- rownames(sce)[
any(grepl("pseudogene", rowData(sce)$ENSEMBL.GENEBIOTYPE))]
Incorporating sample-based annotation
Sarah provided various sample-based metadata about the samples such as the sex, genotype, date of birth, and date of death of each mouse.
Show code
# Add sample metadata
library(readxl)
sample_sheet <- read_excel(
here("data", "sample_sheets", "Minibulk_RNAseq_samples.xlsx"),
sheet = "Sheet1",
range = "A3:I15")
library(janitor)
sample_sheet <- clean_names(sample_sheet)
# Some manual tweaks
sample_sheet$mouse_number <- sub("#", "", sample_sheet$mouse_number)
sample_sheet$genotype.mouse <- factor(
paste0(sample_sheet$smchd1_genotype_updated, ".", sample_sheet$mouse_number))
Sarah sent me a subsequent email (2021-02-22) in which she reported some updated genotyping results, which showed that:
Given the latest genotyping results we have only two male Smchd1-del biological replicates. Both #1269 and #1271 were initially incorrectly genotyped as having the Vav-cre transgene but on repeat genotyping of BM cells we found that they do not, which is why there has been a failure to delete the Smchd1 floxed allele in these samples. This means that #1269 is effectively Smchd1-het and #1271 is WT. After consultation with Marnie, we think it could be useful to include #1269 as a Smchd1-het, although it is lacking the Vav-cre transgene so it may/may not look the same as the other two Smchd1-het males due to this. Also, I’d be keen to know how similar the WT is to the Smchd1-hets. I know there is only one replicate, but we are using the Smchd1-hets as our controls in these experiments, so it would be nice to see how they compare to an actual WT in this context.
These sample-based annotations are included in the colData of the SingleCellExperiment.
Show code
# Join with SCE
colData(sce) <- dplyr::left_join(
as.data.frame(colData(sce)),
sample_sheet,
by = c("mouse_number" = "mouse_number")) %>%
DataFrame(., row.names = colnames(sce))
# Make some key columns factors
sce$smchd1_genotype_updated <- factor(sce$smchd1_genotype_updated)
sce$sex <- factor(sce$sex)
sce$mouse_number <- factor(sce$mouse_number)
sce$genotype.mouse <- factor(
paste0(sce$smchd1_genotype_updated, ".", sce$mouse_number))
# Convoluted way to add tech rep ID
sce$rep <- dplyr::group_by(as.data.frame(colData(sce)), genotype.mouse) %>%
dplyr::mutate(rep = dplyr::row_number()) %>%
dplyr::pull(rep)
colnames(sce) <- paste0(sce$genotype.mouse, ".", sce$rep)
Sarah aimed for technical triplicates of each sample as a 100-cell mini-bulk, but this was not always achievable, as shown in the table below.
Show code
x <- as.data.frame(colData(sce)) %>%
dplyr::count(
smchd1_genotype_updated,
genotyping_bm_cells,
sex,
mouse_number,
dob,
dod,
age_days,
sample_type)
library(gt)
x %>%
dplyr::group_by(smchd1_genotype_updated) %>%
gt(rowname_col = "mouse_number") %>%
tab_spanner(
label = "Sample metadata",
columns = vars(genotyping_bm_cells, sex, dob, dod, age_days)) %>%
tab_header(
title = md("**Samples in C086_Kinkel dataset**"),
subtitle = md("*Rownames are mouse number IDs*")) %>%
tab_source_note(source_note = md("Source: [`data/sample_sheets/Minibulk_RNAseq_samples.xlsx`](../data/sample_sheets/Minibulk_RNAseq_samples.xlsx)")) %>%
summary_rows(
groups = TRUE,
columns = vars(n),
fns = list(total = "sum"),
formatter = fmt_number,
decimals = 0)
| Samples in C086_Kinkel dataset | |||||||
|---|---|---|---|---|---|---|---|
| Rownames are mouse number IDs | |||||||
| Sample metadata | sample_type | n | |||||
| genotyping_bm_cells | sex | dob | dod | age_days | |||
| Del | |||||||
| 1253 | del/del | F | 184/20 | 344/20 | 160 | MiniBulk 100 cells | 3 |
| 1266 | del/del | F | 207/20 | 344/20 | 137 | MiniBulk 100 cells | 2 |
| 1266 | del/del | F | 207/20 | 344/20 | 137 | MiniBulk 58 cells | 1 |
| 1283 | del/del | F | 258/20 | 344/20 | 86 | MiniBulk 100 cells | 1 |
| 1283 | del/del | F | 258/20 | 344/20 | 86 | MiniBulk 50 cells | 1 |
| 1250 | del/del | M | 165/20 | 344/20 | 178 | MiniBulk 100 cells | 3 |
| 1282 | del/del | M | 258/20 | 344/20 | 86 | MiniBulk 100 cells | 3 |
| total | — | — | — | — | — | — | 14 |
| Het | |||||||
| 1264 | del/+ | F | 207/20 | 344/20 | 137 | MiniBulk 100 cells | 3 |
| 1272 | del/+ | F | 221/20 | 344/20 | 123 | MiniBulk 100 cells | 3 |
| 1291 | del/+ | F | 266/20 | 344/20 | 78 | MiniBulk 100 cells | 3 |
| 1248 | del/+ | M | 165/20 | 344/20 | 178 | MiniBulk 100 cells | 3 |
| 1286 | del/+ | M | 258/20 | 344/20 | 86 | MiniBulk 100 cells | 3 |
| 1269 | del/fl | M | 207/20 | 344/20 | 137 | MiniBulk 100 cells | 3 |
| total | — | — | — | — | — | — | 18 |
| WT | |||||||
| 1271 | fl/+ | M | 207/20 | 344/20 | 137 | MiniBulk 100 cells | 3 |
| total | — | — | — | — | — | — | 3 |
Source: data/sample_sheets/Minibulk_RNAseq_samples.xlsx |
|||||||
The complete sample metadata is shown below.
Show code
paged_table(as.data.frame(colData(sce)))
Show code
# Some useful colours
smchd1_genotype_updated_colours <- setNames(
palette.colors(n = nlevels(sce$smchd1_genotype_updated), "Dark 2"),
levels(sce$smchd1_genotype_updated))
sce$smchd1_genotype_updated_colours <- smchd1_genotype_updated_colours[
sce$smchd1_genotype_updated]
sex_colours <- setNames(
RColorBrewer::brewer.pal(4, "Set1")[1:2],
levels(sce$sex))
sce$sex_colours <- sex_colours[sce$sex]
mouse_number_colours <- setNames(
palette.colors(palette = "Alphabet", n = nlevels(sce$mouse_number)),
levels(sce$mouse_number))
sce$mouse_number_colours <- mouse_number_colours[sce$mouse_number]
Have we sequenced enough?
Motivation
SCORE’s mini-bulk protocol is still being finalised and some technical questions remain. In particular, we want to know if we have sequenced enough to answer the biological questions of interest.
One way of estimating if we have sequenced enough uses a metric called ‘sequencing saturation.’ 10x Genomics have some useful information about sequencing saturation available here. In particular,
Sequencing saturation is a measure of the fraction of library complexity that was sequenced in a given experiment. The inverse of the sequencing saturation can be interpreted as the number of additional reads it would take to detect a new transcript. … Sequencing saturation is dependent on the library complexity and sequencing depth. Different cell types will have different amounts of RNA and thus will differ in the total number of different transcripts in the final library (also known as library complexity). … As sequencing depth increases, more genes are detected, but this reaches saturation at different sequencing depths depending on cell type. … Sequencing depth also affects sequencing saturation; generally, the more sequencing reads, the more additional unique transcripts you can detect. However, this is limited by the library complexity.
This naturally leads to the question of ‘how much sequencing saturation should I aim for?’, which very much depends on the goals of the experiment. For this experiment, where we wish to assess differential expression between treatment arms, we will likely want quite high sequencing saturation.
Analysis
To begin, we simply examine the number of reads sequenced/sample.
Figure 4 plots the distribution of the number of reads mapped to exons in each sample3.
Show code
plot(
density(reads_ls),
xlab = expression(log[10](n[reads])),
ylab = "density",
xlim = range(reads_ls),
main = "")
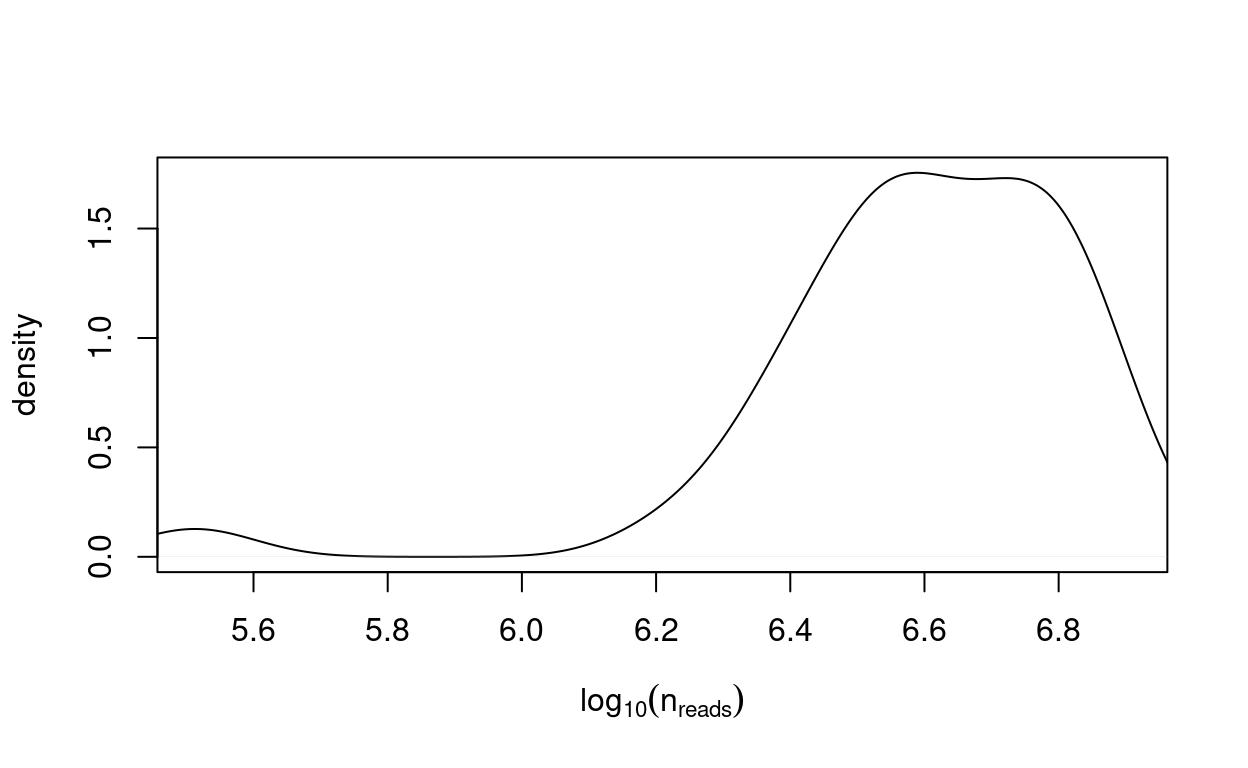
Figure 4: Distribution of the number of reads mapped to exons in each sample.
We estimate the sequencing saturation using 10x Genomics’ formula,
\[ 1 - (n_{deduped reads} / n_{reads}) \] where \(n_{deduped reads}\) = Number of unique (valid cell-barcode, valid UMI, gene)-combinations among confidently mapped reads and \(n_{reads}\) = Total number of confidently mapped, valid cell-barcode, valid UMI reads.
Figure 5 shows that almost all samples are sequenced to near-total saturation.
Show code
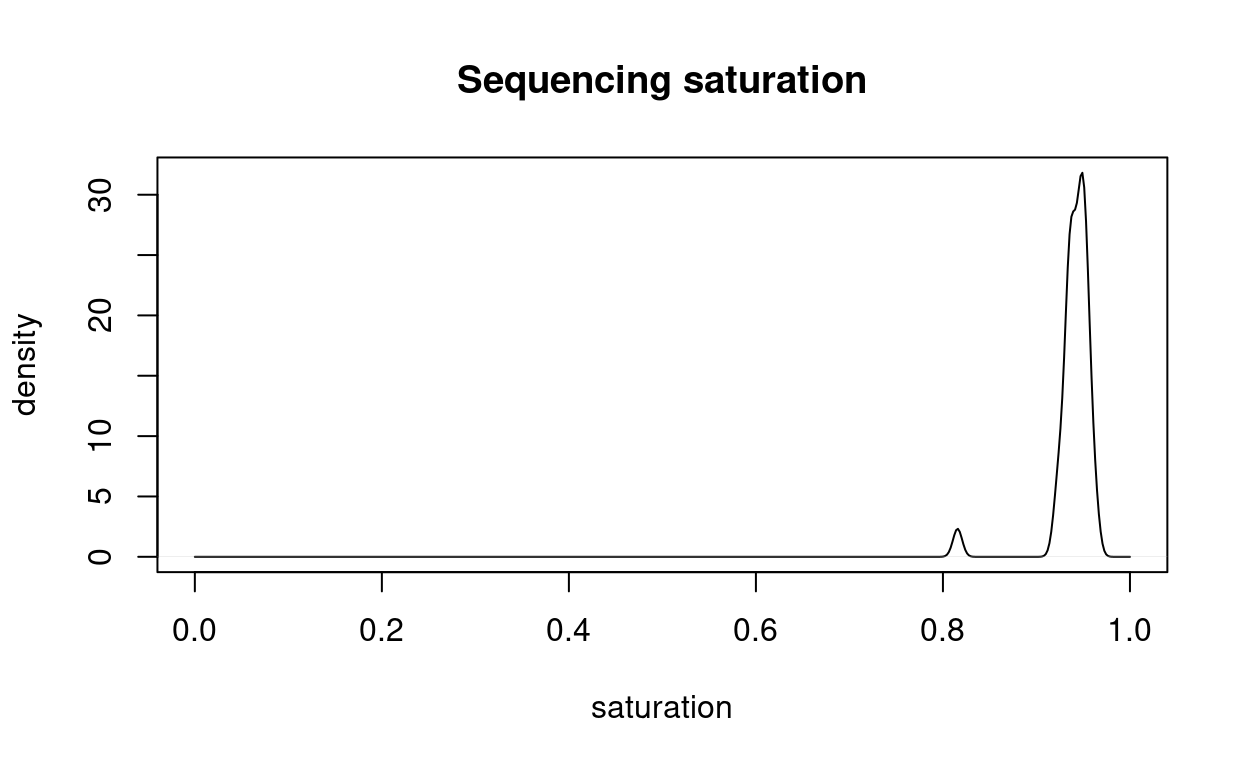
Figure 5: Distribution of the sequencing saturation.
Summary
These results show that the libraries are sequenced to near-saturation.
One way to think of sequencing saturation is provided by 10x Genomics:
The inverse of the sequencing saturation can be interpreted as roughly the number of new transcripts you expect to find with one new read. If sequencing saturation is at 50%, it means that every 2 new reads will result in 1 new UMI count (unique transcript) detected. In contrast, 90% sequencing saturation means that 10 new reads are necessary to obtain one new UMI count. If the sequencing saturation is high, additional sequencing would not recover much new information for the library.
At this stage, it is not necessary to perform additional sequencing of the library.
UMI counts vs. read counts
Motivation
A unique feature of SCORE’s ‘mini-bulk’ RNA-sequencing protocol is the incorporation of unique molecular identifiers (UMIs) into the library preparation; an ordinary bulk RNA-seq protocol does not incorporate UMIs.
UMIs are commonly used in single-cell RNA-sequencing protocols, such as the CEL-Seq2 protocol from which the mini-bulk protocol is adapted. UMIs are molecular tags that are used to detect and quantify unique mRNA transcripts. In this method, mRNA libraries are generated by fragmentation and reverse-transcribed to cDNA. Oligo(dT) primers with specific sequencing linkers are added to the cDNA. Another sequencing linker with a 6 bp random label and an index sequence is added to the 5’ end of the template, which is amplified and sequenced.
The UMI count is always less than or equal to the read count and a UMI count of zero also implies a read count of zero.
Pros of using UMIs:
- Can sequence unique mRNA transcripts
- Can detect transcripts occurring at low frequencies
- Transcripts can be quantified based on sequencing reads specific to each barcode
Cons of using UMIs:
- SCORE’s mini-bulk protocol uses a \(k=6\) bp UMI4 and so (theoretically) the maximum UMI count for each gene is \(4^6 = 4096\).
- There is therefore a risk that highly expressed genes may have their expression measurements artificially truncated, particularly if the sample is deeply sequenced.
To investigate whether expression measurements are being ‘truncated’ in this dataset, and decide whether to use the UMI counts or the read counts, we generated and now analyse two count matrices:
- UMI counts (i.e. deduplicating UMI counts)
- Read counts (i.e. not deduplicating UMI counts, equivalent to ignoring the UMIs)
Analysis
Figure 6 plots the distribution of the logarithm of the read counts. From this we see that the vast majority of read counts are small. In fact:
- 41.4% of read counts are 0
- 95% of the read counts are no greater than 650
- 99% of the read counts are no greater than 2,025
- 99.9% of the read counts are no greater than 11,234
Show code
x <- as.vector(assay(sce, "read_counts")) + 1L
par(mfrow = c(1, 2), cex = 0.5)
plot(
density(log2(x), from = min(log2(x)), to = max(log2(x))),
xlab = expression(log[2](reads + 1)),
main = "",
ylab = "density")
abline(v = log2(4 ^ 6), col = "orange")
plot(
ecdf(log2(x)),
xlim = range(log2(x)),
main = "",
xlab = expression(log[2](reads + 1)),
ylab = "cumulative density")
abline(v = log2(4 ^ 6), col = "orange")
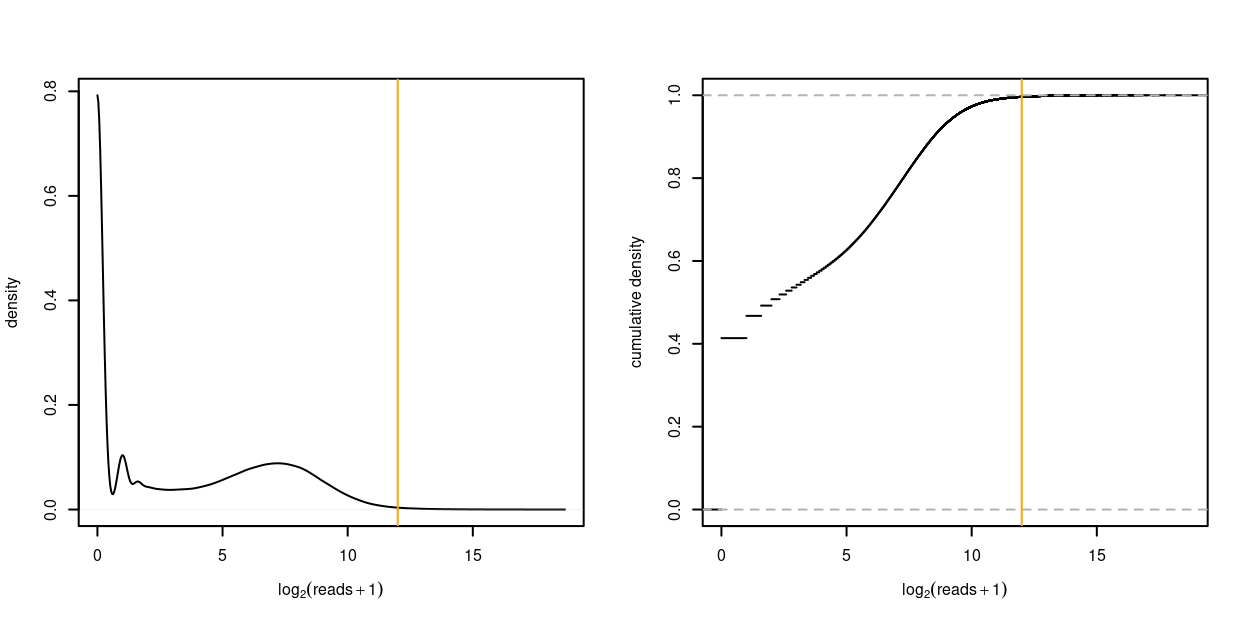
Figure 6: Distribution of the logarithm of the read counts as density plot (left) and empirical cumulative distribution function (right). A count of 1 is added to each UMI count to avoid taking the logarithm of zero. The orange vertical line denotes \(4^6 = 4096\).
Only 0.372% of read counts exceed the \(4^6 = 4096\) threshold. However there are 245 genes for which at least one sample exceeds the \(4^6=4096\) threshold; these genes are shown in the table below. If we were to use the UMI counts, the genes in this list may have their expression measurements artificially truncated in those samples where the number of copies of the transcript exceed the \(4^6=4096\) threshold.
This table shows that many of these genes are mitochondrial and ribosomal protein genes.
Show code
read_e <- as.matrix(assay(sce, "read_counts")[g, ])
umi_e <- as.matrix(assay(sce, "UMI_counts")[g, ])
high_count_tbl <- tibble::tibble(
gene = g,
read_median_count = matrixStats::rowMedians(read_e),
read_exceed_sum = matrixStats::rowSums2(read_e > 4 ^ 6),
read_exceed_percent = read_exceed_sum / ncol(sce),
umi_median_count = matrixStats::rowMedians(umi_e),
umi_exceed_sum = matrixStats::rowSums2(umi_e > 4 ^ 6),
umi_exceed_percent = umi_exceed_sum / ncol(sce)) %>%
dplyr::arrange(desc(read_exceed_sum))
sketch <- htmltools::withTags(table(
class = "display",
thead(
tr(
th(rowspan = 2, "gene"),
th(colspan = 3, "reads"),
th(colspan = 3, "UMIs")
),
tr(
lapply(rep(c("median count", "N > 4096", "% > 4096"), 2), th)
)
)
))
DT::datatable(
high_count_tbl,
caption = "Genes for which any sample has a read count exceeding the 4096 threshold and summaries of the corresponding read and UMI counts, namely: the median count ('median count'); the number of samples for which the counts exceed the threshold ('N > 4096'); and the percentage of samples for which the counts exceed the threshold ('% > 4096').",
container = sketch,
rownames = FALSE) %>%
DT::formatPercentage(
columns = c("read_exceed_percent", "umi_exceed_percent"),
digits = 1) %>%
DT::formatRound(
columns = c("read_median_count", "umi_median_count"),
digits = 0)
We can investigate this list more formally using a statistical test for over-representation of gene ontology (GO) terms in this gene set. This is done using the goana() function from the limma package. These results are shown in the table below and confirm that these genes are enriched for GO terms related to ribosomal and mitochondrial processes. This is something we frequently see with the mini-bulk protocol and the CEL-Seq protocol from which it is derived.
Show code
# NOTE: kegga() was uninformative.
go <- goana(unlist(rowData(sce[g, ])$NCBI.ENTREZID), species = "Mm")
tg <- topGO(go, number = Inf)
DT::datatable(
tg[tg$P.DE < 0.05, ],
caption = "Most significant GO terms for genes with read counts that exceed the 4096 threshold. Report columns are the ontology that the GO term belongs to ('Ont'); the number of genes in the GO term ('N'); the number of genes in the DE set ('DE'); and the p-value for over-representation of the GO term in the set ('P.DE'). The p-values returned are unadjusted for multiple testing because GO terms are often overlapping, so standard methods of p-value adjustment may be very conservative. This means that only very small p-values should be used for published results.") %>%
DT::formatRound(columns = "P.DE", digits = 9)
Figure 7 plots the relationship between UMI counts and read counts for all genes and samples. There are a few notable points about this figure:
- The UMI counts and read counts are very strongly associated.
- The log-fold change between the read counts and the UMI counts can be large, indicating the UMI counts are likely removing substantial PCR amplification noise.
- The log-fold changes between the read counts and the UMI counts are larger on average for more highly expressed genes. The variation in the log-fold change is larger for the lowly expressed genes.
- The hemoglobin genes stand out. These genes are extremely highly expressed, meaning that they likely saturate the 6 bp UMIs and which leads to the strange relationship between the UMI counts and the read counts for these genes.
Show code
x <- log2(as.vector(assay(sce, "UMI_counts")) + 1)
y <- log2(as.vector(assay(sce, "read_counts")) + 1)
m <- (x + y) / 2
d <- y - x
g <- rep(rownames(sce), ncol(sce))
# NOTE: Only showing unique points for plotting speed.
i <- !duplicated(paste0(x, ".", y))
X <- edgeR::cpm(assay(sce, "UMI_counts"), log = TRUE)
Y <- edgeR::cpm(assay(sce, "read_counts"), log = TRUE)
M <- (X + Y) / 2
D <- y - x
G <- g
# NOTE: Only showing unique points for plotting speed.
I <- !duplicated(paste0(x, ".", y))
par(mfrow = c(2, 2))
plotWithHighlights(
x = x[i],
y = y[i],
status = ifelse(g[i] %in% ribo_set, 1, ifelse(g[i] %in% mito_set, -1, 0)),
legend = FALSE,
bg.pch = ".",
hl.pch = ".",
hl.col = c("orange", "dodgerBlue"),
hl.cex = 3,
sub = paste0("r = ", number(cor(x, y), accuracy = 0.001)),
xlab = expression(log[2](UMIs + 1)),
ylab = expression(log[2](reads + 1)))
abline(a = 0, b = 1, lty = 2, col = "grey")
plotWithHighlights(
x = m[i] / 2,
y = d[i],
pch = ".",
status = ifelse(g[i] %in% ribo_set, 1, ifelse(g[i] %in% mito_set, -1, 0)),
legend = FALSE,
bg.pch = ".",
hl.pch = ".",
hl.col = c("orange", "dodgerBlue"),
hl.cex = 3,
xlab = "Average log-(count+1)",
ylab = "logFC (reads vs. UMIs)",
ylim = c(-max(abs(c(d, D))), max(abs(c(d, D)))))
abline(h = 0, lty = 2, col = "grey")
l <- lowess(m, d, f = 0.3)
lines(l, col = "red", lwd = 2)
plotWithHighlights(
x = X[I],
y = Y[I],
status = ifelse(G[I] %in% ribo_set, 1, ifelse(G[I] %in% mito_set, -1, 0)),
legend = FALSE,
bg.pch = ".",
hl.pch = ".",
hl.col = c("orange", "dodgerBlue"),
hl.cex = 3,
sub = paste0(
"r = ",
number(cor(as.vector(X), as.vector(Y)), accuracy = 0.001)),
xlab = expression(logCPM(UMIs)),
ylab = expression(logCPM(reads)))
abline(a = 0, b = 1, lty = 2, col = "grey")
plotWithHighlights(
x = M[I],
y = D[i],
status = ifelse(G[I] %in% ribo_set, 1, ifelse(G[I] %in% mito_set, -1, 0)),
legend = FALSE,
bg.pch = ".",
hl.pch = ".",
hl.col = c("orange", "dodgerBlue"),
hl.cex = 3,
xlab = "Average logCPM",
ylab = "logFC (reads vs. UMIs)",
ylim = c(-max(abs(c(d, D))), max(abs(c(d, D)))))
abline(h = 0, lty = 2, col = "grey")
l <- lowess(M, D, f = 0.3)
lines(l, col = "red", lwd = 2)
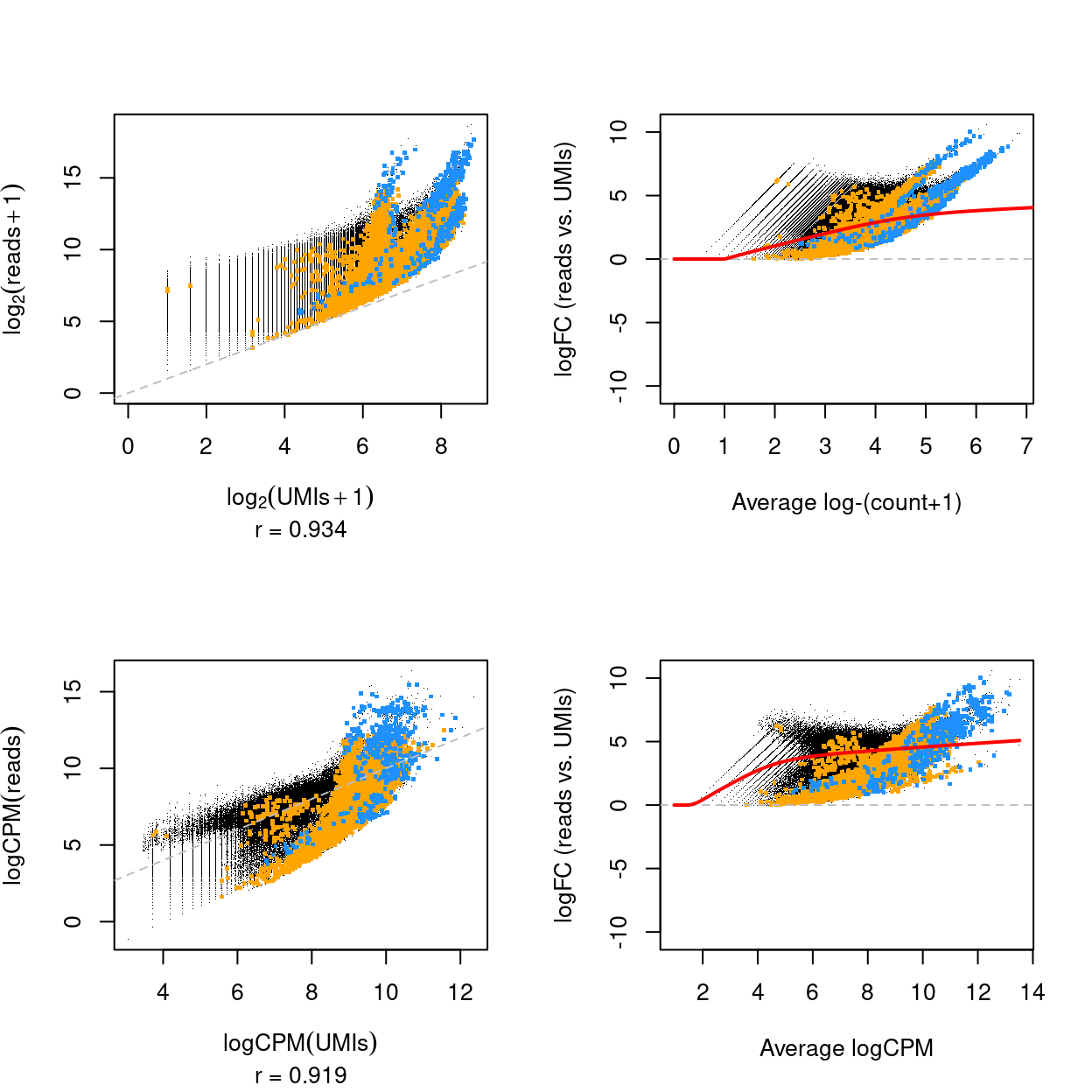
Figure 7: Scatterplot of the logarithm of the UMI and read counts (top) and logCPM of the UMI and read counts (bottom) for all genes and samples (left) and mean-difference plot of the same data (right). The correlation in the scatterplot is reported below the x-axis label. Genes that are ribosomal protein genes subunits are highlighted in orange and mitochondrial genes in blue. The red line is a trend fitted to the mean-differences.
Rather than looking at all samples and all genes at once, can also examine the relationship between UMI counts and read counts within a single sample across genes (Figure 8) and within a single gene across samples (Figure 9).
Show code
X <- edgeR::cpm(assay(sce, "UMI_counts"), log = TRUE)
Y <- edgeR::cpm(assay(sce, "read_counts"), log = TRUE)
cor_j <- diag(cor(X, Y))
# NOTE: Splitting up the cor() computations is a hack to improve speed whilst
# keeping memory usage somewhat under control.
b <- 1000
s <- seq(1, by = b, to = nrow(sce))
e <- pmin(s + b - 1, nrow(sce))
cor_i <- unlist(
Map(
f = function(ss, ee ) diag(cor(t(X[ss:ee, ]), t(Y[ss:ee, ]))),
ss = s,
ee = e))
These figures show that the UMI counts and read counts are highly correlated across both samples (median = 0.916, median absolute deviation = 0.006) and genes (median = 0.808, median absolute deviation = 0.208).
Show code
par(mfrow = c(3, 3), cex = 0.5)
jj <- names(sort(cor_j)[c(1:3, 17:18, 33:35)])
for (j in jj) {
g <- rownames(sce)
plot(
x = X[, j],
y = Y[, j],
pch = ".",
cex = ifelse(g %in% c(ribo_set, mito_set), 5, 1),
col = ifelse(
g %in% ribo_set, "orange",
ifelse(g %in% mito_set, "dodgerBlue", "black")),
sub = paste0("r = ", number(cor_j[j], accuracy = 0.001)),
xlab = "logCPM(UMIs)",
ylab = "logCPM(reads)",
main = j,
xlim = range(x),
ylim = range(y))
abline(a = 0, b = 1, col = "grey", lty = 2)
}
plot(
density(cor_j, na.rm = TRUE, from = -1, to = 1),
main = "All samples",
xlab = expression(r[p]))
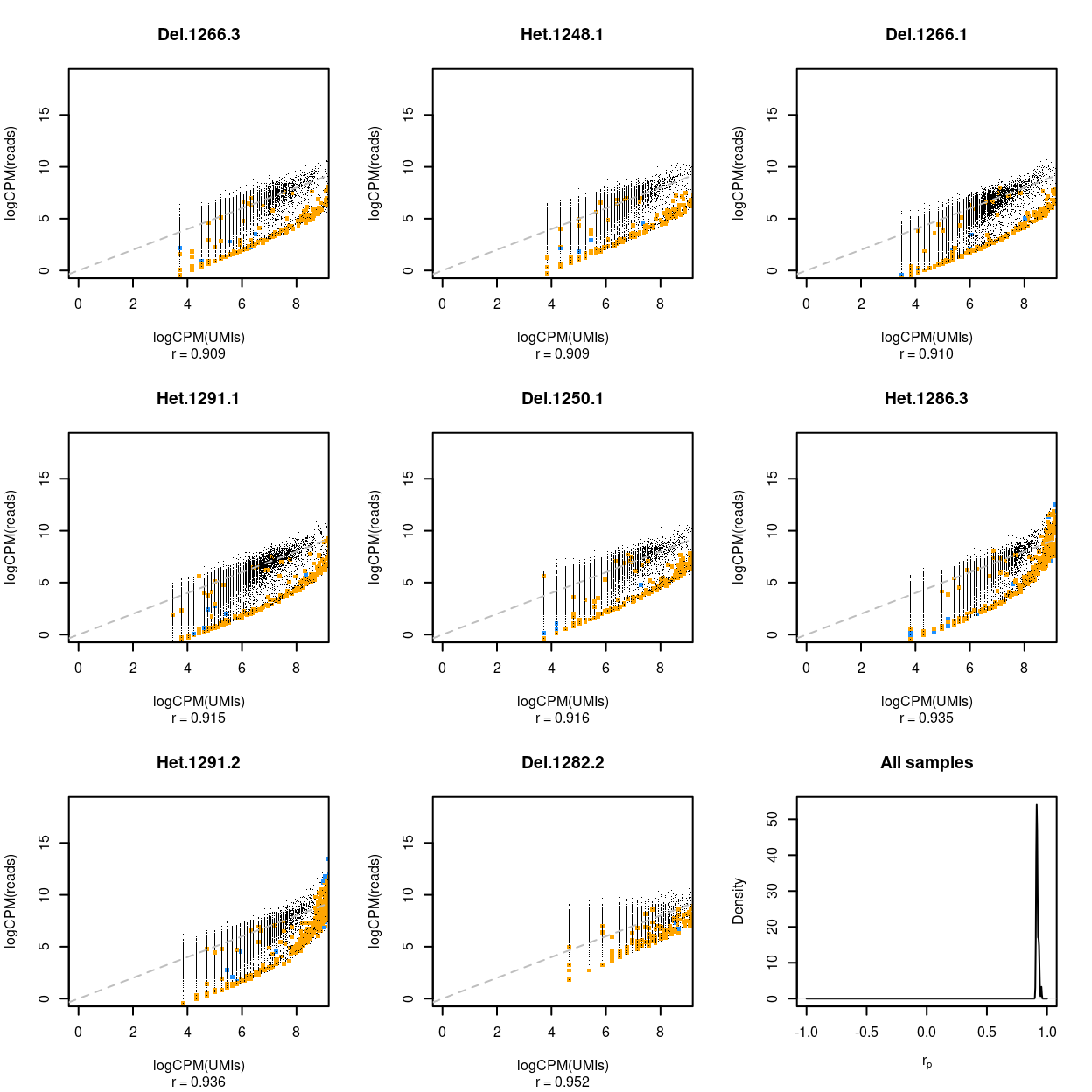
Figure 8: Association between UMI counts and read counts for each gene within each sample. The first 8 panels are scatter plots of the UMI counts and read counts for each gene for the samples with the 3 lowest correlations, 2 middlemost correlations, and 3 highest correlations. The correlation in the scatterplot is reported below each panel. Genes that are ribosomal protein genes subunits are highlighted in orange and mitochondrial genes in blue. The final panel is the distribution of these correlations across all samples.
Show code
par(mfrow = c(3, 3), cex = 0.5)
# NOTE: There are some NA correlations, so this changes ii.
ii <- names(sort(cor_i)[c(1:3, 12792:12793, 25585:25587)])
for (i in ii) {
g <- i
plot(
x = X[i, ],
y = Y[i, ],
pch = 16,
col = ifelse(
g %in% ribo_set, "orange",
ifelse(g %in% mito_set, "dodgerBlue", "black")),
sub = paste0("r = ", number(cor_i[i], accuracy = 0.001)),
xlab = "logCPM(UMIs)",
ylab = "logCPM(reads)",
main = i,
xlim = range(X),
ylim = range(Y))
abline(a = 0, b = 1, col = "grey", lty = 2)
}
plot(
density(cor_i, na.rm = TRUE, from = -1, to = 1),
main = "All genes",
xlab = expression(r[p]))
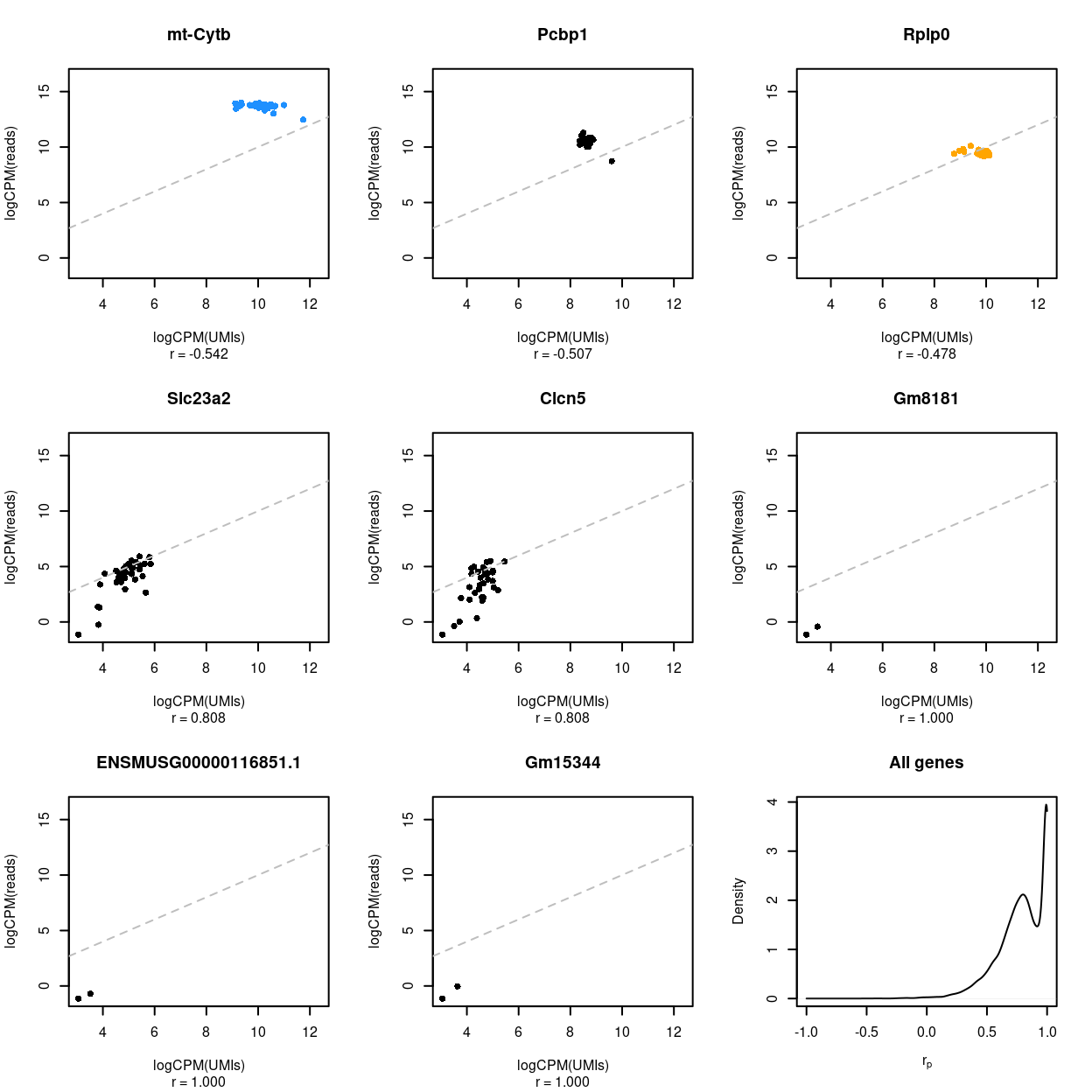
Figure 9: Association between UMI counts and read counts for each sample within each gene. The first 8 panels are scatter plots of the UMI counts and read counts for each sample for the genes with the 3 lowest correlations, 2 middlemost correlations, and 3 highest correlations. The correlation in the scatterplot is reported below each panel. Genes that are ribosomal protein genes subunits are highlighted in orange and mitochondrial genes in blue. The final panel is the distribution of these correlations across all genes.
Summary
The ultimate aim of this analysis is to perform a differential expression (DE) analysis. On the one hand, a DE analysis is more powerful when there are more counts available, which is where the read counts are favoured. On the other hand, a DE analysis may be biased if these counts arise through PCR duplicates, which is where the deduplicated UMI counts are favoured. The choice between using the read counts or the UMI counts in the DE analysis is therefore a trade-off between these two choices.
For now, we defer our choice of read counts vs. UMI counts, but the remainder of this preprocessing report uses read counts.
Quality control of samples
Defining the quality control metrics
Low-quality samples need to be removed to ensure that technical effects do not distort downstream analysis results. We use several quality control (QC) metrics to measure the quality of the samples:
sum: This measures the library size of the samples, which is the total sum of counts across genes. We want samples to have high library sizes as this means more RNA has been successfully captured during library preparation.detected: This is the number of expressed features5 in each sample. Samples with few expressed features are likely to be of poor quality, as the diverse transcript population has not been successful captured.subsets_Mt_percent: This measures the proportion of reads which are mapped to mitochondrial RNA. If there is a higher than expected proportion of mitochondrial RNA this is often symptomatic of a sample which is under stress and is therefore of low quality and will not be used for the analysis.subsets_Ribo_percent: This measures the proportion of reads which are mapped to ribosomal protein genes. If there is a higher than expected proportion of ribosomal protein gene expression this is often symptomatic of a cell which is of compromised quality and we may want to exclude it from the analysis.
For mini-bulk data, we typically observe library sizes that are in the hundreds of thousands6 and several thousand expressed genes per cell.
Show code
is_mito <- rownames(sce) %in% mito_set
summary(is_mito)
is_ribo <- rownames(sce) %in% ribo_set
summary(is_ribo)
sce_reads <- addPerCellQC(
sce,
subsets = list(Mt = which(is_mito), Ribo = which(is_ribo)),
assay.type = "read_counts",
use.altexps = FALSE)
sce_umis <- addPerCellQC(
sce,
subsets = list(Mt = which(is_mito), Ribo = which(is_ribo)),
assay.type = "UMI_counts",
use.altexps = FALSE)
The aim is to remove putative low-quality samples that have low library sizes, low numbers of expressed features, and high mitochondrial proportions. Such samples can interfere with downstream analyses, e.g., by forming distinct clusters that complicate interpretation of the results.
Visualizing the QC metrics
The distributions of these metrics are shown in Figure 10 for read and UMI counts.
Show code
p1 <- plotColData(
sce_reads,
"sum",
x = "genotype.mouse",
colour_by = "smchd1_genotype_updated") +
scale_y_log10() +
theme_cowplot(font_size = 6) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
scale_y_log10() +
annotation_logticks(
sides = "l",
short = unit(0.03, "cm"),
mid = unit(0.06, "cm"),
long = unit(0.09, "cm")) +
scale_colour_manual(values = smchd1_genotype_updated_colours)
p2 <- plotColData(
sce_reads,
"detected",
x = "genotype.mouse",
colour_by = "smchd1_genotype_updated") +
theme_cowplot(font_size = 6) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
scale_colour_manual(values = smchd1_genotype_updated_colours)
p3 <- plotColData(
sce_reads,
"subsets_Mt_percent",
x = "genotype.mouse",
colour_by = "smchd1_genotype_updated") +
theme_cowplot(font_size = 6) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
scale_colour_manual(values = smchd1_genotype_updated_colours)
p4 <- plotColData(
sce_reads,
"subsets_Ribo_percent",
x = "genotype.mouse",
colour_by = "smchd1_genotype_updated") +
theme_cowplot(font_size = 6) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
scale_colour_manual(values = smchd1_genotype_updated_colours)
q1 <- p1 + p2 + p3 + p4 + plot_layout(ncol = 4)
p5 <- plotColData(
sce_umis,
"sum",
x = "genotype.mouse",
colour_by = "smchd1_genotype_updated") +
scale_y_log10() +
theme_cowplot(font_size = 6) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
scale_y_log10() +
annotation_logticks(
sides = "l",
short = unit(0.03, "cm"),
mid = unit(0.06, "cm"),
long = unit(0.09, "cm")) +
scale_colour_manual(values = smchd1_genotype_updated_colours)
p6 <- plotColData(
sce_umis,
"detected",
x = "genotype.mouse",
colour_by = "smchd1_genotype_updated") +
theme_cowplot(font_size = 6) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
scale_colour_manual(values = smchd1_genotype_updated_colours)
p7 <- plotColData(
sce_umis,
"subsets_Mt_percent",
x = "genotype.mouse",
colour_by = "smchd1_genotype_updated") +
theme_cowplot(font_size = 6) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
scale_colour_manual(values = smchd1_genotype_updated_colours)
p8 <- plotColData(
sce_umis,
"subsets_Ribo_percent",
x = "genotype.mouse",
colour_by = "smchd1_genotype_updated") +
theme_cowplot(font_size = 6) +
theme(axis.text.x = element_text(angle = 45, hjust = 1)) +
scale_colour_manual(values = smchd1_genotype_updated_colours)
q2 <- p5 + p6 + p7 + p8 + plot_layout(ncol = 4)
q1 / q2 +
plot_layout(guides = "collect") +
plot_annotation(
title = "QC metrics",
subtitle = "Reads counts (top row) and UMI counts (bottom row)")
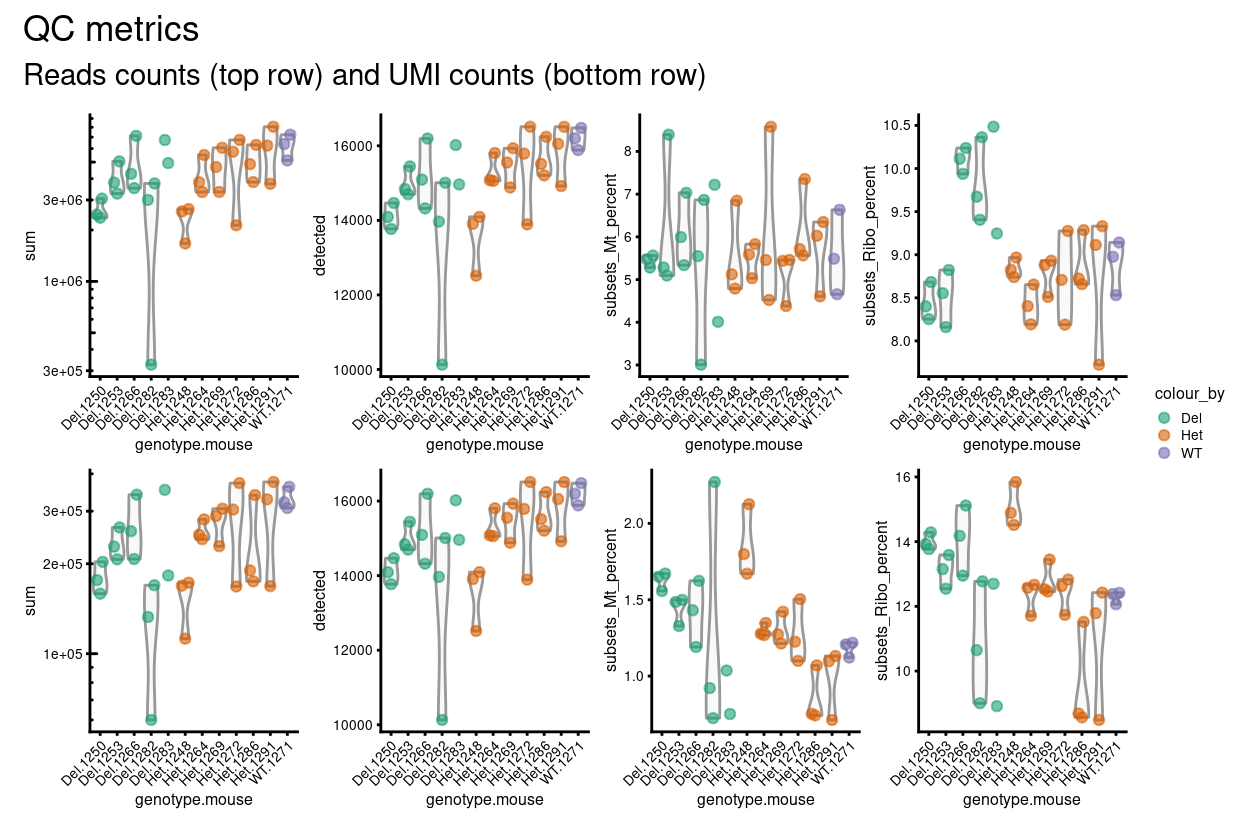
Figure 10: Distributions of various QC metrics for all samples in the data set. This includes the library sizes, number of expressed genes, and proportion of reads mapped to mitochondrial genes.
Identifying outliers by each metric
Outliers are defined based on the median absolute deviation (MADs) from the median value of each metric across all samples.
Show code
libsize_drop_reads <- isOutlier(
metric = sce_reads$sum,
nmads = 3,
type = "lower",
log = TRUE)
libsize_drop_umis <- isOutlier(
metric = sce_umis$sum,
nmads = 3,
type = "lower",
log = TRUE)
feature_drop_reads <- isOutlier(
metric = sce_reads$detected,
nmads = 3,
type = "lower",
log = TRUE)
feature_drop_umis <- isOutlier(
metric = sce_umis$detected,
nmads = 3,
type = "lower",
log = TRUE)
The following table summarises the QC cutoffs:
Show code
libsize_drop_df <- data.frame(
reads_lower = attributes(libsize_drop_reads)$thresholds["lower"],
umis_lower = attributes(libsize_drop_umis)$thresholds["lower"])
feature_drop_df <- data.frame(
reads_lower = attributes(feature_drop_reads)$thresholds["lower"],
umis_lower = attributes(feature_drop_umis)$thresholds["lower"])
qc_cutoffs_df <- cbind(libsize_drop_df, feature_drop_df)
colnames(qc_cutoffs_df) <- c(
"read counts", "umi counts", "read features", "umi features")
qc_cutoffs_df %>%
knitr::kable(caption = "QC cutoffs", digits = 0)
| read counts | umi counts | read features | umi features | |
|---|---|---|---|---|
| lower | 757058 | 63235 | 11974 | 11974 |
We would typically remove outliers (e.g., small outliers for the library size and the number of expressed features) at this stage. However, we opt to retain all samples for the time being. The following table highlights samples that would typically exclude as outliers by this criteria:
Show code
keep <- !(libsize_drop_reads | libsize_drop_umis | feature_drop_reads | feature_drop_umis)
x <- cbind(
data.frame(sample = colnames(sce), keep = keep),
colData(sce_reads)[, c("sum", "detected")],
colData(sce_umis)[, c("sum", "detected")])
colnames(x) <- c(
colnames(x)[1:2],
paste0("reads_", colnames(x)[3:4]),
paste0("umis_", colnames(x)[5:6]))
row.names(x) <- NULL
DT::datatable(
x[order(keep), ],
caption = "Quality control metrics including library size (`sum`) and number of genes detected (`keep`) for each replicate. Each replicate is annotated by whether it would be kept or excluded by the typical quality control procedures (`keep`).",
rownames = FALSE)
Summary
We have opted not to exclude any replicates at this stage. However, we have identified 1 replicate (Del.1282.2) that has unusually small library size (by reads and UMIs) and number of genes detected compared to the other samples. We will keep an eye on this replicate and may later remove it.
Examining gene-level metrics
Inspecting the most highly expressed genes
Figures 11 shows the most highly expressed genes in the dataset. Many of these genes are mitochondrial genes and ribosomal protein.
Show code
plotHighestExprs(sce, n = 50, exprs_values = "read_counts") +
ggtitle("Read counts") +
plotHighestExprs(sce, n = 50, exprs_values = "UMI_counts") +
ggtitle("UMI counts")
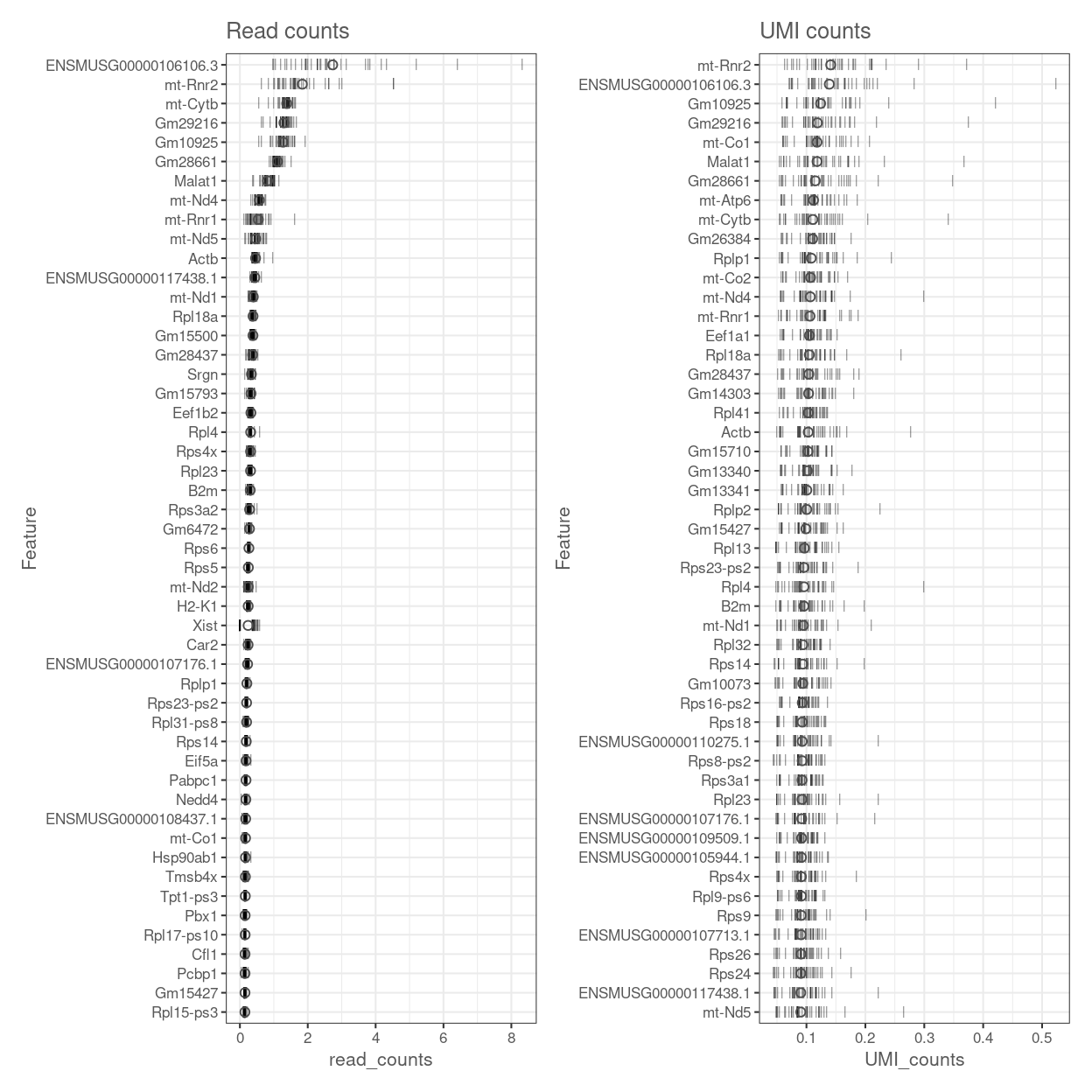
Figure 11: Percentage of total counts assigned to the top 50 most highly-abundant features in the data set. For each feature, each bar represents the percentage assigned to that feature for a single sample, while the circle represents the average across all samples. Bars are coloured by the total number of expressed features in each samples, while circles are coloured according to whether the feature is labelled as a control feature.
Figure 12 shows the most highly expressed after excluding the mitochondrial genes and ribosomal protein.
Show code
plotHighestExprs(
sce,
drop_features = intersect(rownames(sce), c(mito_set, ribo_set)),
n = 50,
exprs_values = "read_counts") +
ggtitle("Read counts") +
plotHighestExprs(
sce,
drop_features = intersect(rownames(sce), c(mito_set, ribo_set)),
n = 50,
exprs_values = "UMI_counts") +
ggtitle("UMI counts")
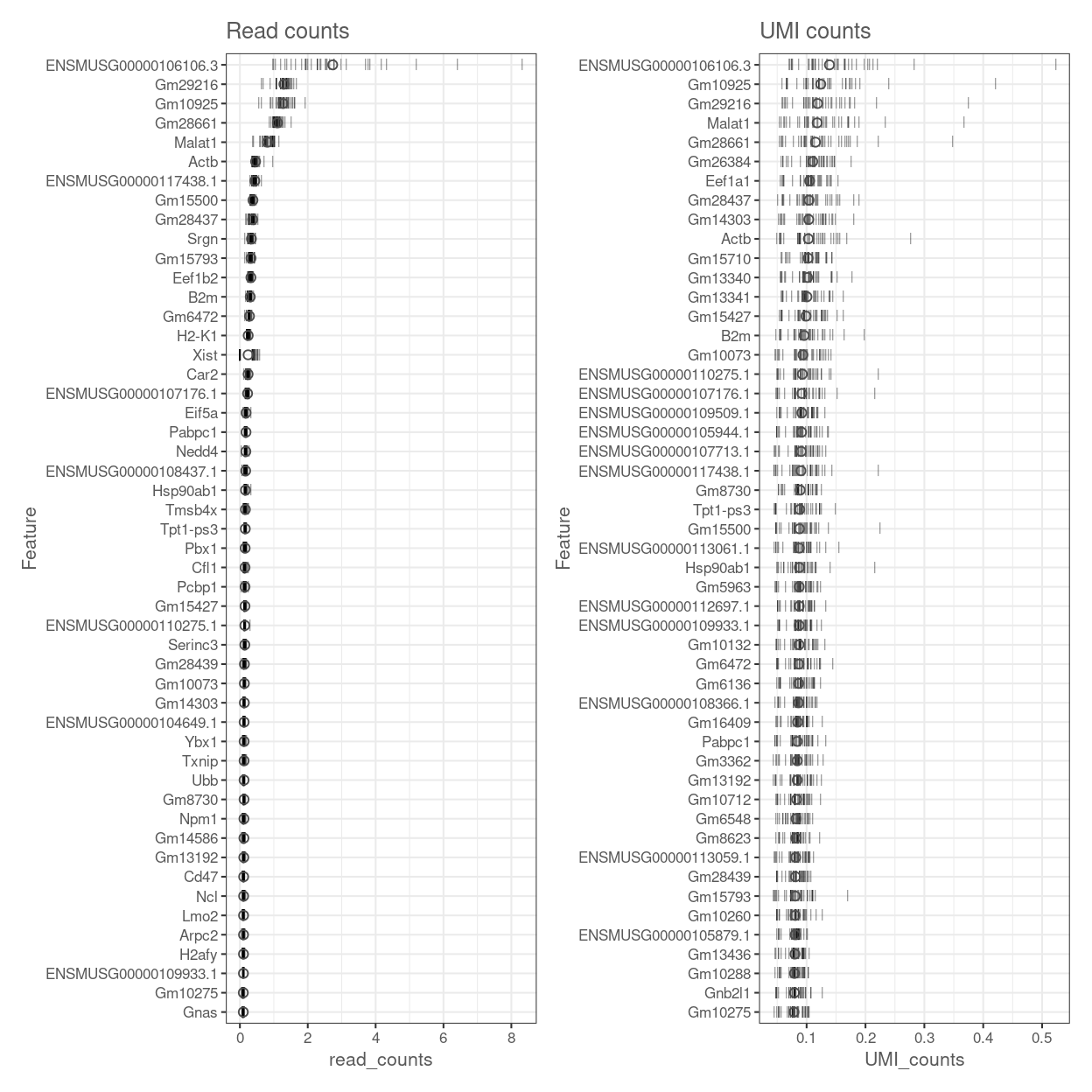
Figure 12: Percentage of total counts assigned to the top 50 most highly-abundant features (after excluding mitochondrial genes and ribosomal protein genes) in the data set. For each feature, each bar represents the percentage assigned to that feature for a single sample, while the circle represents the average across all samples. Bars are coloured by the total number of expressed features in each sample, while circles are coloured according to whether the feature is labelled as a control feature.
Sex check using Xist
Figure 13 visualises the expression of Xist, a female-specific gene. This can be used to confirm the reported sex of the sample and to detect sample mix-ups. Figure 13 shows that female samples have much higher average expression of Xist than male samples, as we would expect.
There is one replicate, Del.1282.2, that has is nominally male but has \(logCPM(UMI_{counts})\) for Xist that is comparable to the female samples. However, we have already seen that this replicate has an unusually small library size (by reads and UMIs) and number of genes detected compared to the other samples, so it already suspect.
Show code
assay(sce, "logCPM_reads") <- edgeR::cpm(assay(sce, "read_counts"), log = TRUE)
assay(sce, "logCPM_umis") <- edgeR::cpm(assay(sce, "UMI_counts"), log = TRUE)
p1 <- plotExpression(
sce,
"Xist",
x = "sex",
exprs_values = "read_counts",
colour_by = "sex") +
scale_colour_manual(values = sex_colours, name = "sex") +
guides(fill = FALSE) +
ylab("Read counts") +
ylim(0, NA)
p2 <-plotExpression(
sce,
"Xist",
x = "sex",
exprs_values = "logCPM_reads",
colour_by = "sex") +
scale_colour_manual(values = sex_colours, name = "sex") +
guides(fill = FALSE) +
ylab("logCPM(read counts)")
p3 <- plotExpression(
sce,
"Xist",
x = "sex",
exprs_values = "UMI_counts",
colour_by = "sex") +
scale_colour_manual(values = sex_colours, name = "sex") +
guides(fill = FALSE) +
ylab("UMI counts") +
ylim(0, NA)
p4 <-plotExpression(
sce,
"Xist",
x = "sex",
exprs_values = "logCPM_umis",
colour_by = "sex") +
scale_colour_manual(values = sex_colours, name = "sex") +
guides(fill = FALSE) +
ylab("logCPM(UMI counts)")
p1 + p2 + p3 + p4 + plot_layout(guides = "collect", ncol = 2)
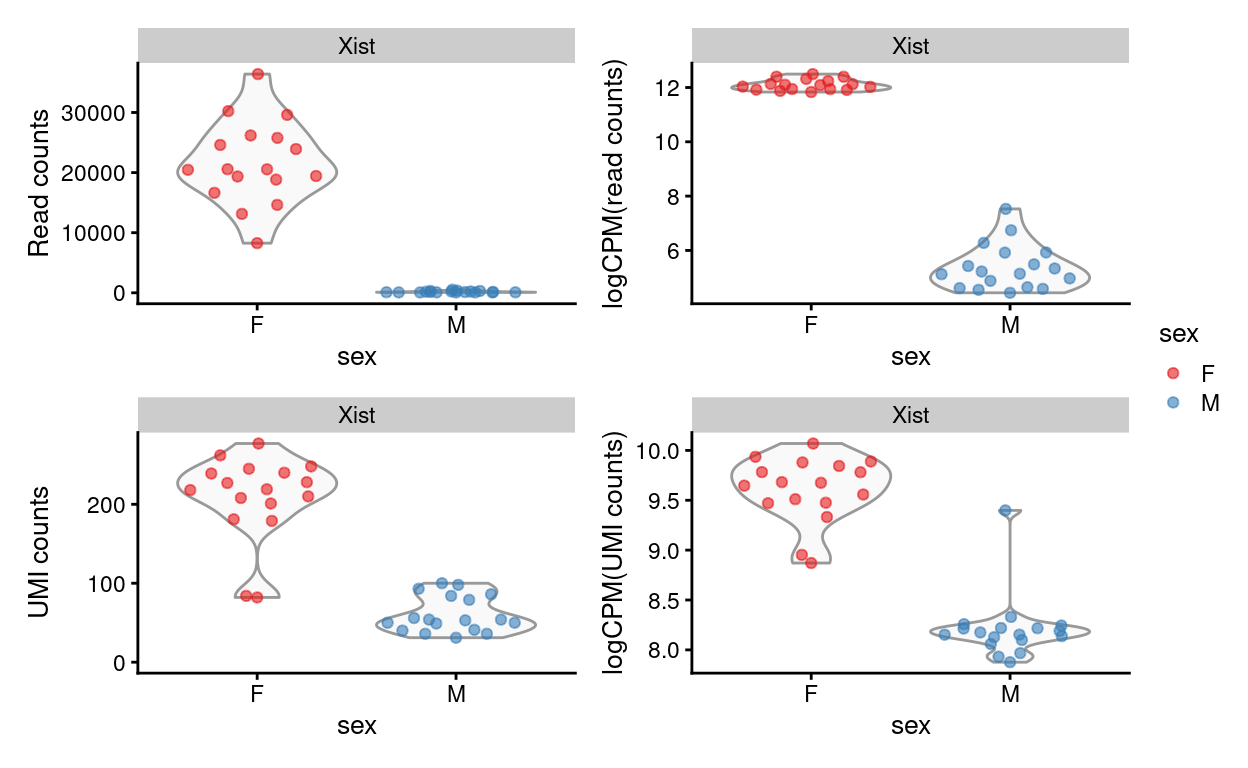
Figure 13: Expression of Xist by mouse on a ‘raw counts’ and ‘logCPM’ scale using the read or UMI counts.
Genotype check using Smchd1
Figure 14 visualises the expression of Smchd1, the gene under investigation and which was deleted via a floxed allele in the Del mice. This can be used to confirm the reported genotype of the sample and to detect sample mix-ups. The Del samples have somewhat lower average expression of Smchd1 than Het and WT samples on, as we would expect, but the difference is not a striking on-off binary difference. Furthermore, the Del samples have positive counts of Smchd1.
Show code
p1 <- plotExpression(
sce,
"Smchd1",
x = "genotype.mouse",
exprs_values = "read_counts",
colour_by = "smchd1_genotype_updated") +
scale_colour_manual(
values = smchd1_genotype_updated_colours,
name = "smchd1_genotype_updated") +
guides(fill = FALSE) +
ylab("Read counts") +
scale_x_discrete(guide = guide_axis(angle = 45)) +
ylim(0, NA)
p2 <- plotExpression(
sce,
"Smchd1",
x = "genotype.mouse",
exprs_values = "logCPM_reads",
colour_by = "smchd1_genotype_updated") +
scale_colour_manual(
values = smchd1_genotype_updated_colours,
name = "smchd1_genotype_updated") +
guides(fill = FALSE) +
ylab("logCPM(read counts)") +
scale_x_discrete(guide = guide_axis(angle = 45))
p3 <- plotExpression(
sce,
"Smchd1",
x = "genotype.mouse",
exprs_values = "UMI_counts",
colour_by = "smchd1_genotype_updated") +
scale_colour_manual(
values = smchd1_genotype_updated_colours,
name = "smchd1_genotype_updated") +
guides(fill = FALSE) +
ylab("UMI counts") +
scale_x_discrete(guide = guide_axis(angle = 45)) +
ylim(0, NA)
p4 <- plotExpression(
sce,
"Smchd1",
x = "genotype.mouse",
exprs_values = "logCPM_umis",
colour_by = "smchd1_genotype_updated") +
scale_colour_manual(
values = smchd1_genotype_updated_colours,
name = "smchd1_genotype_updated") +
guides(fill = FALSE) +
ylab("logCPM(UMI counts)") +
scale_x_discrete(guide = guide_axis(angle = 45))
p1 + p2 + p3 + p4 + plot_layout(guides = "collect", ncol = 2)
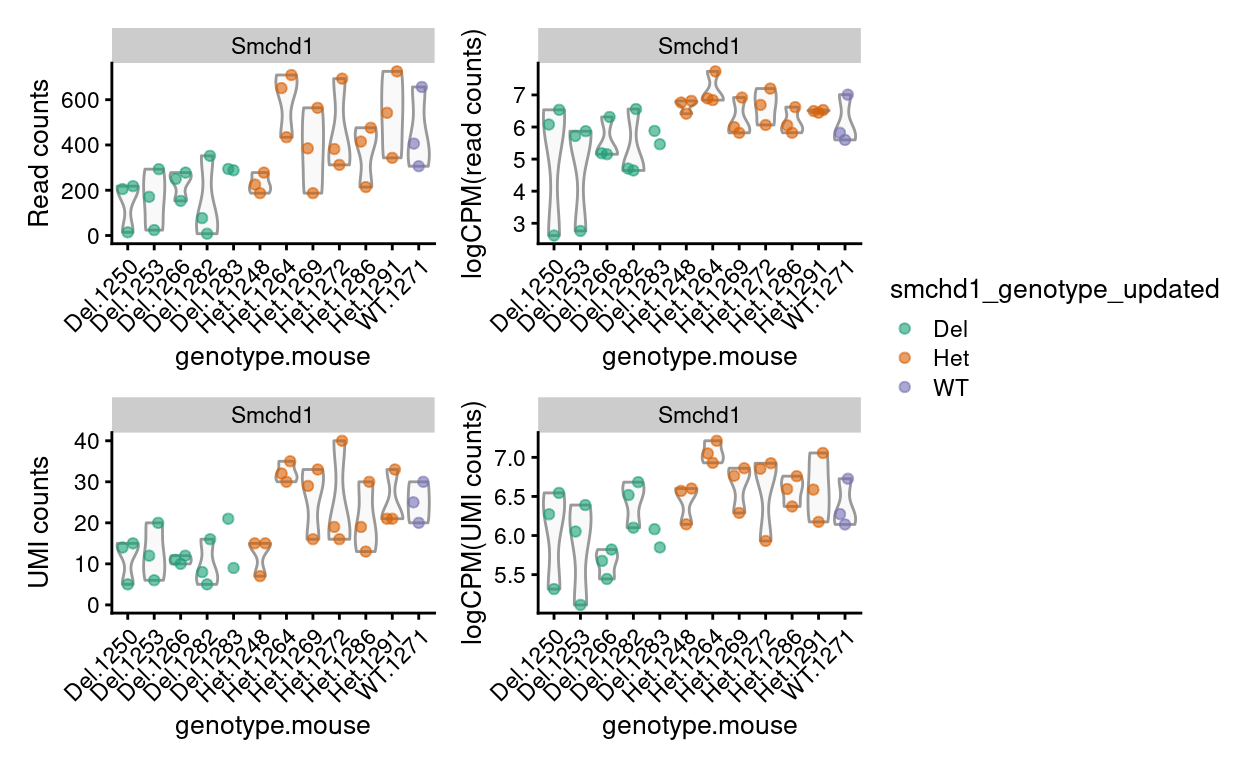
Figure 14: Expression of Smchd1 by mouse on a ‘raw counts’ and ‘logCPM’ scale using the read or UMI counts.
Concluding remarks
The processed SingleCellExperiment object is available (see data/SCEs/C086_Kinkel.preprocessed.SCE.rds). This will be used in downstream analyses, i.e. differential expression analysis.
Additional information
The following are available on request:
- Full CSV tables of any data presented.
- PDF/PNG files of any static plots.
Session info
Show code
sessioninfo::session_info()
─ Session info ─────────────────────────────────────────────────────
setting value
version R version 4.0.3 (2020-10-10)
os CentOS Linux 7 (Core)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_US.UTF-8
ctype en_US.UTF-8
tz Australia/Melbourne
date 2021-10-15
─ Packages ─────────────────────────────────────────────────────────
! package * version date lib
P AnnotationDbi * 1.52.0 2020-10-27 [?]
P AnnotationFilter * 1.14.0 2020-10-27 [?]
P askpass 1.1 2019-01-13 [?]
P assertthat 0.2.1 2019-03-21 [?]
P backports 1.2.1 2020-12-09 [?]
P beachmat 2.6.4 2020-12-20 [?]
P beeswarm 0.2.3 2016-04-25 [?]
P Biobase * 2.50.0 2020-10-27 [?]
P BiocFileCache 1.14.0 2020-10-27 [?]
P BiocGenerics * 0.36.0 2020-10-27 [?]
P BiocManager 1.30.10 2019-11-16 [?]
P BiocNeighbors 1.8.2 2020-12-07 [?]
P BiocParallel * 1.24.1 2020-11-06 [?]
P BiocSingular 1.6.0 2020-10-27 [?]
P BiocStyle 2.18.1 2020-11-24 [?]
P biomaRt 2.46.3 2021-02-09 [?]
P Biostrings 2.58.0 2020-10-27 [?]
P bit 4.0.4 2020-08-04 [?]
P bit64 4.0.5 2020-08-30 [?]
P bitops 1.0-6 2013-08-17 [?]
P blob 1.2.1 2020-01-20 [?]
P cachem 1.0.4 2021-02-13 [?]
P cellranger 1.1.0 2016-07-27 [?]
P checkmate 2.0.0 2020-02-06 [?]
P cli 2.3.1 2021-02-23 [?]
P colorspace 2.0-0 2020-11-11 [?]
P commonmark 1.7 2018-12-01 [?]
P cowplot * 1.1.1 2020-12-30 [?]
P crayon 1.4.1 2021-02-08 [?]
P crosstalk 1.1.1 2021-01-12 [?]
P curl 4.3 2019-12-02 [?]
P DBI 1.1.1 2021-01-15 [?]
P dbplyr 2.1.0 2021-02-03 [?]
P DelayedArray 0.16.1 2021-01-22 [?]
P DelayedMatrixStats 1.12.3 2021-02-03 [?]
P DEoptimR 1.0-8 2016-11-19 [?]
P digest 0.6.27 2020-10-24 [?]
P distill 1.2 2021-01-13 [?]
P downlit 0.2.1 2020-11-04 [?]
P dplyr * 1.0.4 2021-02-02 [?]
P DT 0.17 2021-01-06 [?]
P edgeR 3.32.1 2021-01-14 [?]
P ellipsis 0.3.1 2020-05-15 [?]
P EnsDb.Mmusculus.v79 * 2.99.0 2020-07-15 [?]
P ensembldb * 2.14.0 2020-10-27 [?]
P evaluate 0.14 2019-05-28 [?]
P fansi 0.4.2 2021-01-15 [?]
P farver 2.0.3 2020-01-16 [?]
P fastmap 1.1.0 2021-01-25 [?]
P generics 0.1.0 2020-10-31 [?]
P GenomeInfoDb * 1.26.2 2020-12-08 [?]
P GenomeInfoDbData 1.2.4 2020-10-20 [?]
P GenomicAlignments 1.26.0 2020-10-27 [?]
P GenomicFeatures * 1.42.1 2020-11-12 [?]
P GenomicRanges * 1.42.0 2020-10-27 [?]
P GGally 2.1.0 2021-01-06 [?]
P ggbeeswarm 0.6.0 2017-08-07 [?]
P ggplot2 * 3.3.3 2020-12-30 [?]
P glue 1.4.2 2020-08-27 [?]
P GO.db * 3.12.1 2020-11-04 [?]
P graph 1.68.0 2020-10-27 [?]
P gridExtra 2.3 2017-09-09 [?]
P gt * 0.2.2 2020-08-05 [?]
P gtable 0.3.0 2019-03-25 [?]
P here * 1.0.1 2020-12-13 [?]
P highr 0.8 2019-03-20 [?]
P hms 1.0.0 2021-01-13 [?]
P htmltools 0.5.1.1 2021-01-22 [?]
P htmlwidgets 1.5.3 2020-12-10 [?]
P httr 1.4.2 2020-07-20 [?]
P IRanges * 2.24.1 2020-12-12 [?]
P irlba 2.3.3 2019-02-05 [?]
P janitor * 2.1.0 2021-01-05 [?]
P jsonlite 1.7.2 2020-12-09 [?]
P knitr 1.31 2021-01-27 [?]
P labeling 0.4.2 2020-10-20 [?]
P lattice 0.20-41 2020-04-02 [3]
P lazyeval 0.2.2 2019-03-15 [?]
P lifecycle 1.0.0 2021-02-15 [?]
P limma * 3.46.0 2020-10-27 [?]
P locfit 1.5-9.4 2020-03-25 [?]
P lubridate 1.7.9.2 2020-11-13 [?]
P magrittr 2.0.1 2020-11-17 [?]
P Matrix 1.2-18 2019-11-27 [3]
P MatrixGenerics * 1.2.1 2021-01-30 [?]
P matrixStats * 0.58.0 2021-01-29 [?]
P mclust 5.4.7 2020-11-20 [?]
P memoise 2.0.0 2021-01-26 [?]
P msigdbr * 7.2.1 2020-10-02 [?]
P munsell 0.5.0 2018-06-12 [?]
P Mus.musculus * 1.3.1 2020-07-15 [?]
P openssl 1.4.3 2020-09-18 [?]
P org.Hs.eg.db 3.12.0 2020-10-20 [?]
P org.Mm.eg.db * 3.12.0 2020-10-20 [?]
P OrganismDbi * 1.32.0 2020-10-27 [?]
P patchwork * 1.1.1 2020-12-17 [?]
P pillar 1.5.0 2021-02-22 [?]
P pkgconfig 2.0.3 2019-09-22 [?]
P plyr 1.8.6 2020-03-03 [?]
P prettyunits 1.1.1 2020-01-24 [?]
P progress 1.2.2 2019-05-16 [?]
P ProtGenerics 1.22.0 2020-10-27 [?]
P purrr 0.3.4 2020-04-17 [?]
P R6 2.5.0 2020-10-28 [?]
P rappdirs 0.3.3 2021-01-31 [?]
P RBGL 1.66.0 2020-10-27 [?]
P RColorBrewer 1.1-2 2014-12-07 [?]
P Rcpp 1.0.6 2021-01-15 [?]
P RCurl 1.98-1.2 2020-04-18 [?]
P readxl * 1.3.1 2019-03-13 [?]
P rematch 1.0.1 2016-04-21 [?]
P reshape 0.8.8 2018-10-23 [?]
P Rhtslib 1.22.0 2020-10-27 [?]
P rlang 0.4.10 2020-12-30 [?]
P rmarkdown * 2.11 2021-09-14 [?]
P robustbase 0.93-7 2021-01-04 [?]
P rprojroot 2.0.2 2020-11-15 [?]
P Rsamtools 2.6.0 2020-10-27 [?]
P RSQLite 2.2.3 2021-01-24 [?]
P rsvd 1.0.3 2020-02-17 [?]
P rtracklayer 1.50.0 2020-10-27 [?]
P S4Vectors * 0.28.1 2020-12-09 [?]
P sass 0.3.1 2021-01-24 [?]
P scales * 1.1.1 2020-05-11 [?]
P scater * 1.18.5 2021-02-16 [?]
P scPipe * 1.12.0 2020-10-27 [?]
P scuttle 1.0.4 2020-12-17 [?]
P sessioninfo 1.1.1 2018-11-05 [?]
P SingleCellExperiment * 1.12.0 2020-10-27 [?]
P snakecase 0.11.0 2019-05-25 [?]
P sparseMatrixStats 1.2.1 2021-02-02 [?]
P stringi 1.5.3 2020-09-09 [?]
P stringr 1.4.0 2019-02-10 [?]
P SummarizedExperiment * 1.20.0 2020-10-27 [?]
P tibble 3.0.6 2021-01-29 [?]
P tidyr * 1.1.2 2020-08-27 [?]
P tidyselect 1.1.0 2020-05-11 [?]
P TxDb.Mmusculus.UCSC.mm10.knownGene * 3.10.0 2020-07-15 [?]
P utf8 1.1.4 2018-05-24 [?]
P vctrs 0.3.6 2020-12-17 [?]
P vipor 0.4.5 2017-03-22 [?]
P viridis 0.5.1 2018-03-29 [?]
P viridisLite 0.3.0 2018-02-01 [?]
P withr 2.4.1 2021-01-26 [?]
P xfun 0.26 2021-09-14 [?]
P XML 3.99-0.5 2020-07-23 [?]
P xml2 1.3.2 2020-04-23 [?]
P XVector 0.30.0 2020-10-27 [?]
P yaml 2.2.1 2020-02-01 [?]
P zlibbioc 1.36.0 2020-10-27 [?]
source
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.0)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.2)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.2)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.2)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
Bioconductor
[1] /stornext/Projects/score/Analyses/C086_Kinkel/renv/library/R-4.0/x86_64-pc-linux-gnu
[2] /tmp/RtmpwbxEJA/renv-system-library
[3] /stornext/System/data/apps/R/R-4.0.3/lib64/R/library
P ── Loaded and on-disk path mismatch.In the terminology of Bioconductor this is called a SingleCellExperiment object.↩︎
Some care is taken to account for missing and duplicate gene symbols; missing symbols are replaced with the Ensembl identifier and duplicated symbols are concatenated with the (unique) Ensembl identifiers.↩︎
I.e. the ‘library size’ when using read counts↩︎
The choice of a 6 bp UMI in the mini-bulk protocol is because it is adapted from SCORE’s CEL-Seq2 single-cell RNA-sequencing protocol. With scRNA-seq data it is very rare to observe more than \(4^k = 4^6 = 4096\) unique molecules for any one gene.↩︎
The number of expressed genes refers to the number of genes which have non-zero counts (ie. they have been identified in the sample at least once)↩︎
This is consistent with the use of UMI counts rather than read counts, as each transcript molecule can only produce one UMI count but can yield many reads after fragmentation.↩︎