Setup
Show code
library(SingleCellExperiment)
library(here)
library(cowplot)
library(patchwork)
library(edgeR)
library(scater)
library(scran)
sce <- readRDS(here("data", "SCEs", "C057_Cooney.annotated.SCE.rds"))
cycling_sce <- readRDS(
here("data", "SCEs", "C057_Cooney.cycling.annotated.SCE.rds"))
not_cycling_sce <- readRDS(
here("data", "SCEs", "C057_Cooney.not_cycling.annotated.SCE.rds"))
# Some useful colours
sample_colours <- setNames(
unique(sce$sample_colours),
unique(names(sce$sample_colours)))
treatment_colours <- setNames(
unique(sce$treatment_colours),
unique(names(sce$treatment_colours)))
cluster_colours <- setNames(
unique(sce$cluster_colours),
unique(names(sce$cluster_colours)))
# Re-level Treatment
sce$Treatment <- relevel(sce$Treatment, "Uninfected")
cycling_sce$Treatment <- relevel(cycling_sce$Treatment, "Uninfected")
not_cycling_sce$Treatment <- relevel(not_cycling_sce$Treatment, "Uninfected")
source(here("code", "helper_functions.R"))
# BCL family members provided by James
bcl2_df <- read.csv(here("data/group-1057.csv"))
Analysis ignoring cycling_subset
The results of this analysis are as if we had done a bulk RNA-seq experiment rather than scRNA-seq. It ignores the within-sample heterogeneity and consequently we recommend the results from the other differential analyses.
There is no DA analysis when ignoring cycling_subset.
DE
We require that a gene has an absolute fold change (\(FC\)) \(>1.5\) (i.e. \(|log_2(FC)| > log_2(1.5) \approx 0.58\)) and an \(FDR < 0.05\) to be called as differentially expressed. This is achieved by using the glmTreat() method in edgeR, which is a statistically rigorous method for thresholded differential expression testing.
Please see output/DEGs/cycling_subset/ for heatmaps and spreadsheets of these DEG lists, including results of GO and KEGG analyses using the goana() and kegga() functions from the limma package. The heatmaps show up to the top-50 DEGs (ordered by \(FDR\)).
This directory also contains interactive Glimma plots of the differential expression results. The Glimma plots show the pseudobulk data for that label.
Show code
# NOTE: Have to drop the complicated TRA and TRB DFrameList objects.
x <- sce
x$TRA <- NULL
x$TRB <- NULL
sce.ignoring_cycling_subset <- aggregateAcrossCells(
x,
id = colData(x)[, c("Sample", "Treatment")],
coldata.merge = FALSE,
use.dimred = FALSE,
use.altexps = FALSE)
sizeFactors(sce.ignoring_cycling_subset) <- NULL
sce.ignoring_cycling_subset <- logNormCounts(sce.ignoring_cycling_subset)
colLabels(sce.ignoring_cycling_subset) <- "ignoring_cycling_subset"
colnames(sce.ignoring_cycling_subset) <-
paste0(colLabels(sce.ignoring_cycling_subset),
".",
sce.ignoring_cycling_subset$Sample)
Show code
Show code
sce.ignoring_cycling_subset_filt <-
sce.ignoring_cycling_subset[, sce.ignoring_cycling_subset$ncells >= 10]
de_results <- pseudoBulkDGE(
sce.ignoring_cycling_subset_filt,
label = sce.ignoring_cycling_subset_filt$label,
design = ~Treatment,
coef = "TreatmentInfected",
condition = sce.ignoring_cycling_subset_filt$Treatment,
include.intermediates = TRUE,
lfc = log2(1.5))
- Labels for which we couldn’t run DE (
character(0)means none)
Show code
metadata(de_results)$failed
character(0)- Number of DEGs per label (
-1means downregulated inInfectedvs.Uninfected,0means not DE,1means upregulated inInfectedvs.Uninfected)
Show code
is.de <- decideTestsPerLabel(de_results, threshold = 0.05)
summarizeTestsPerLabel(is.de)
-1 0 1 NA
ignoring_cycling_subset 74 11562 92 8874Show code
outdir <- here("output", "DEGs", "ignoring_cycling_subset")
dir.create(outdir, recursive = TRUE)
createDEGOutputs(
outdir = outdir,
de_results = de_results,
summed_filt = sce.ignoring_cycling_subset_filt,
sce = sce,
fdr = 0.05)
Show code
lapply(names(de_results), function(n) {
se <- sce.ignoring_cycling_subset_filt[
, sce.ignoring_cycling_subset_filt$label == n]
x <- de_results[[n]]
x <- x[order(x$FDR), ]
features <- rownames(x[which(x$FDR < 0.05), ])
if (length(features) > 2) {
plotHeatmap(
se,
features = head(features, 50),
center = TRUE,
zlim = c(-3, 3),
symmetric = TRUE,
color = hcl.colors(101, "Blue-Red 3"),
order_columns_by = c("Treatment", "Sample"),
column_annotation_colors = list(
Treatment = treatment_colours,
Sample = sample_colours),
cluster_rows = TRUE,
fontsize = 8,
main = n,
filename = file.path(outdir, paste0(n, ".pseudobulk_heatmap.pdf")))
plotGroupedHeatmap(
sce,
features = head(features, 50),
group = "Sample",
center = TRUE,
symmetric = TRUE,
color = hcl.colors(101, "Blue-Red 3"),
cluster_rows = TRUE,
fontsize = 8,
main = n,
cluster_cols = FALSE,
filename = file.path(
outdir,
paste0(n, ".average_expression_heatmap.pdf")))
}
})
Gene set testing
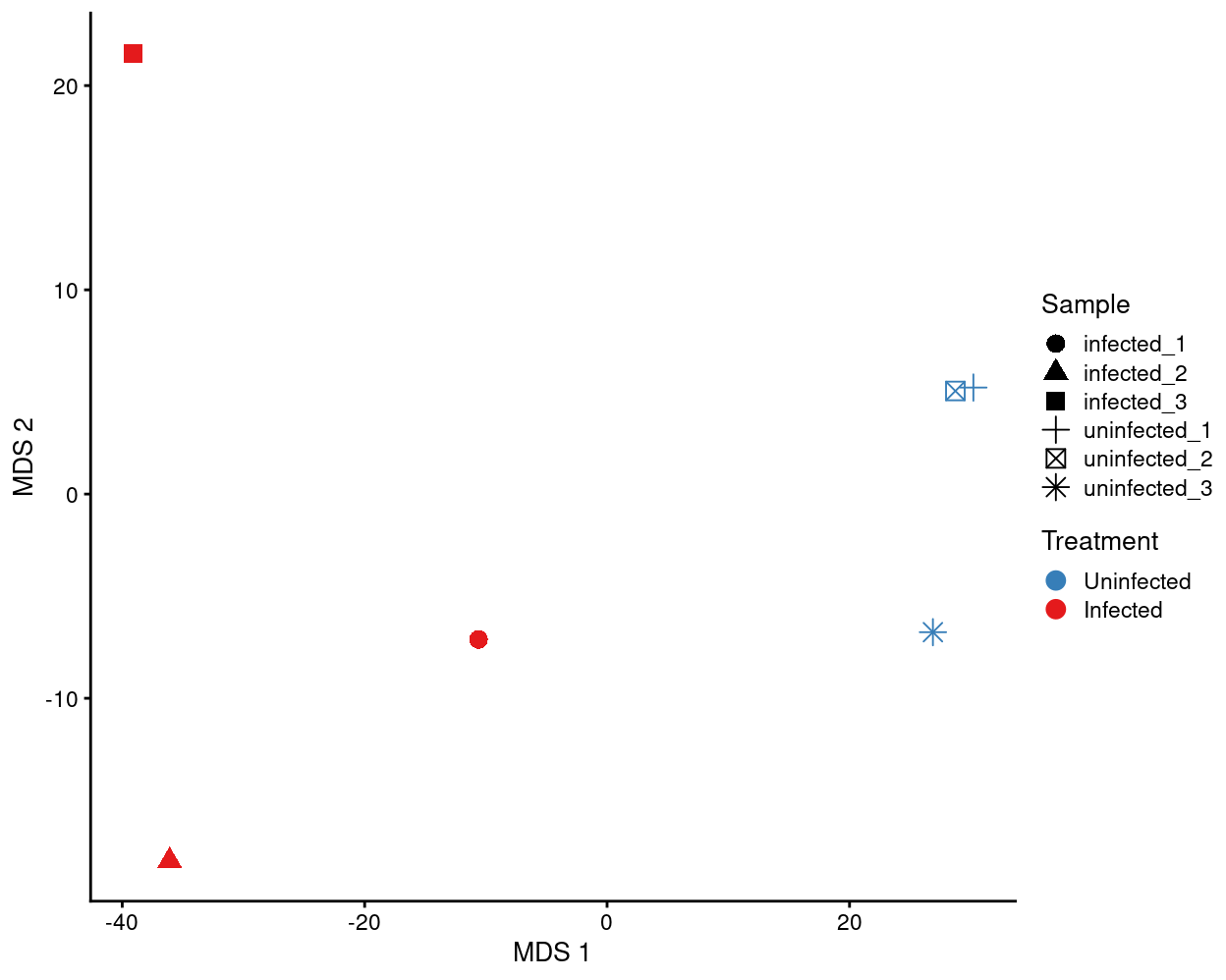
We use the fry() function from the edgeR R/Bioconductor package to perform a self-contained gene set test against the null hypothesis that none of the genes in the BCL-family gene set (supplied by James) are differentially expressed. We can visualise the expression of the genes in any given set and in any given comparison by highlighting those genes on the MD plot and/or using a ‘barcode plot.’ Such figures are often included in publications.
Show code
# NOTE: The following is a workaround to the lack of support for tabsets in
# distill (see https://github.com/rstudio/distill/issues/11 and
# https://github.com/rstudio/distill/issues/11#issuecomment-692142414 in
# particular).
xaringanExtra::use_panelset()
ignoring_cycling_subset
Show code
n <- "ignoring_cycling_subset"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 20 | Down | 0.0624809 | 0.000225 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
Analysis based on cycling_subset
DE
We require that a gene has an absolute fold change (\(FC\)) \(>1.5\) (i.e. \(|log_2(FC)| > log_2(1.5) \approx 0.58\)) and an \(FDR < 0.05\) to be called as differentially expressed. This is achieved by using the glmTreat() method in edgeR, which is a statistically rigorous method for thresholded differential expression testing.
Please see output/DEGs/cycling_subset/ for heatmaps and spreadsheets of these DEG lists, including results of GO and KEGG analyses using the goana() and kegga() functions from the limma package. The heatmaps show up to the top-50 DEGs (ordered by \(FDR\)).
This directory also contains interactive Glimma plots of the differential expression results. The Glimma plots show the pseudobulk data for that label.
Show code
# NOTE: Have to drop the complicated TRA and TRB DFrameList objects.
x <- sce
x$TRA <- NULL
x$TRB <- NULL
sce.cycling_subset_Sample <- aggregateAcrossCells(
x,
id = colData(x)[, c("cycling_subset", "Sample", "Treatment")],
coldata.merge = FALSE,
use.dimred = FALSE,
use.altexps = FALSE)
sizeFactors(sce.cycling_subset_Sample) <- NULL
sce.cycling_subset_Sample <- logNormCounts(sce.cycling_subset_Sample)
colLabels(sce.cycling_subset_Sample) <- sce.cycling_subset_Sample$cycling_subset
colnames(sce.cycling_subset_Sample) <-
paste0(sce.cycling_subset_Sample$cycling_subset,
".",
sce.cycling_subset_Sample$Sample)
Show code
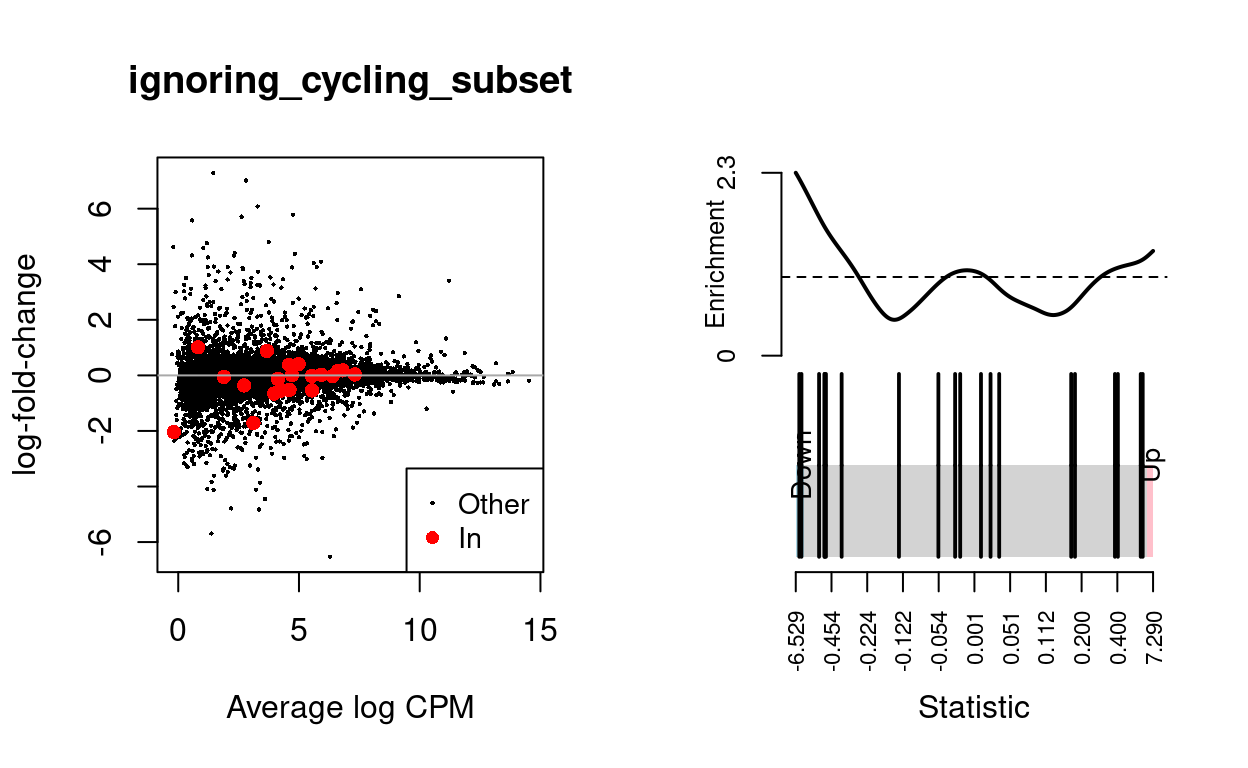
sce.cycling_subset_Sample <- runMDS(sce.cycling_subset_Sample)
p1 <- plotMDS(
sce.cycling_subset_Sample,
colour_by = "Treatment",
shape_by = "cycling_subset",
point_size = 3,
point_alpha = 2) +
scale_colour_manual(values = treatment_colours, name = "Treatment")
p2 <- plotMDS(
sce.cycling_subset_Sample,
colour_by = "Sample",
shape_by = "cycling_subset",
point_size = 3,
point_alpha = 2) +
scale_colour_manual(values = sample_colours, name = "Sample")
p1 + p2
Show code
sce.cycling_subset_Sample_filt <-
sce.cycling_subset_Sample[, sce.cycling_subset_Sample$ncells >= 10]
de_results <- pseudoBulkDGE(
sce.cycling_subset_Sample_filt,
label = sce.cycling_subset_Sample_filt$cycling_subset,
design = ~Treatment,
coef = "TreatmentInfected",
condition = sce.cycling_subset_Sample_filt$Treatment,
include.intermediates = TRUE,
lfc = log2(1.5))
- Labels for which we couldn’t run DE (
character(0)means none)
Show code
metadata(de_results)$failed
character(0)- Number of DEGs per label (
-1means downregulated inInfectedvs.Uninfected,0means not DE,1means upregulated inInfectedvs.Uninfected)
Show code
is.de <- decideTestsPerLabel(de_results, threshold = 0.05)
summarizeTestsPerLabel(is.de)
-1 0 1 NA
Cycling 38 9799 53 10712
Not cycling 82 11016 97 9407Show code
outdir <- here("output", "DEGs", "cycling_subset")
dir.create(outdir, recursive = TRUE)
createDEGOutputs(
outdir = outdir,
de_results = de_results,
summed_filt = sce.cycling_subset_Sample_filt,
sce = sce,
fdr = 0.05)
Show code
lapply(names(de_results), function(n) {
se <- sce.cycling_subset_Sample_filt[, sce.cycling_subset_Sample_filt$label == n]
x <- de_results[[n]]
x <- x[order(x$FDR), ]
features <- rownames(x[which(x$FDR < 0.05), ])
if (length(features) > 2) {
plotHeatmap(
se,
features = head(features, 50),
center = TRUE,
zlim = c(-3, 3),
symmetric = TRUE,
color = hcl.colors(101, "Blue-Red 3"),
order_columns_by = c("Treatment", "Sample"),
column_annotation_colors = list(
Treatment = treatment_colours,
Sample = sample_colours),
cluster_rows = TRUE,
fontsize = 8,
main = n,
filename = file.path(outdir, paste0(n, ".pseudobulk_heatmap.pdf")))
plotGroupedHeatmap(
sce,
features = head(features, 50),
group = "Sample",
columns = sce$cycling_subset == n,
center = TRUE,
symmetric = TRUE,
color = hcl.colors(101, "Blue-Red 3"),
cluster_rows = TRUE,
fontsize = 8,
main = n,
cluster_cols = FALSE,
filename = file.path(
outdir,
paste0(n, ".average_expression_heatmap.pdf")))
}
})
Gene set testing
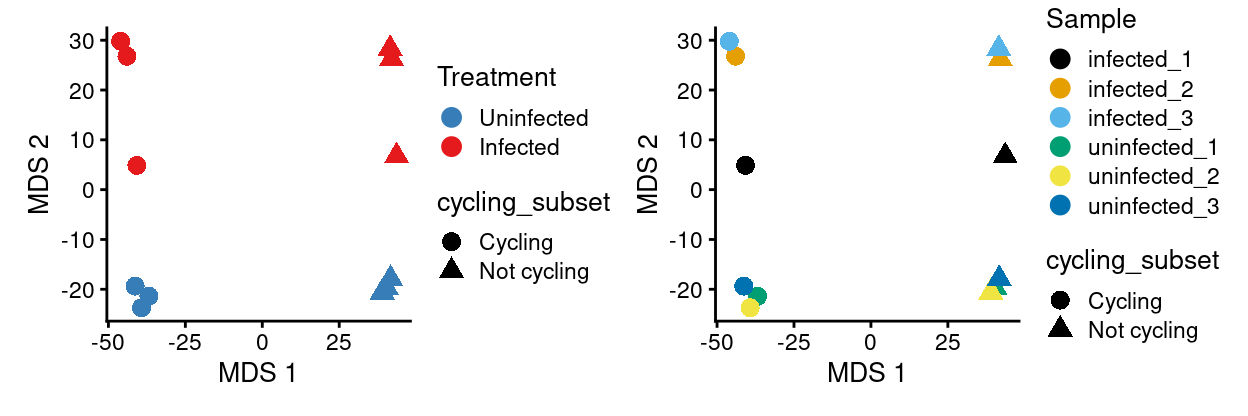
We use the fry() function from the edgeR R/Bioconductor package to perform a self-contained gene set test against the null hypothesis that none of the genes in the BCL-family gene set (supplied by James) are differentially expressed. We can visualise the expression of the genes in any given set and in any given comparison by highlighting those genes on the MD plot and/or using a ‘barcode plot.’ Such figures are often included in publications.
Cycling
Show code
n <- "Cycling"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 17 | Down | 0.5963644 | 0.0048845 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
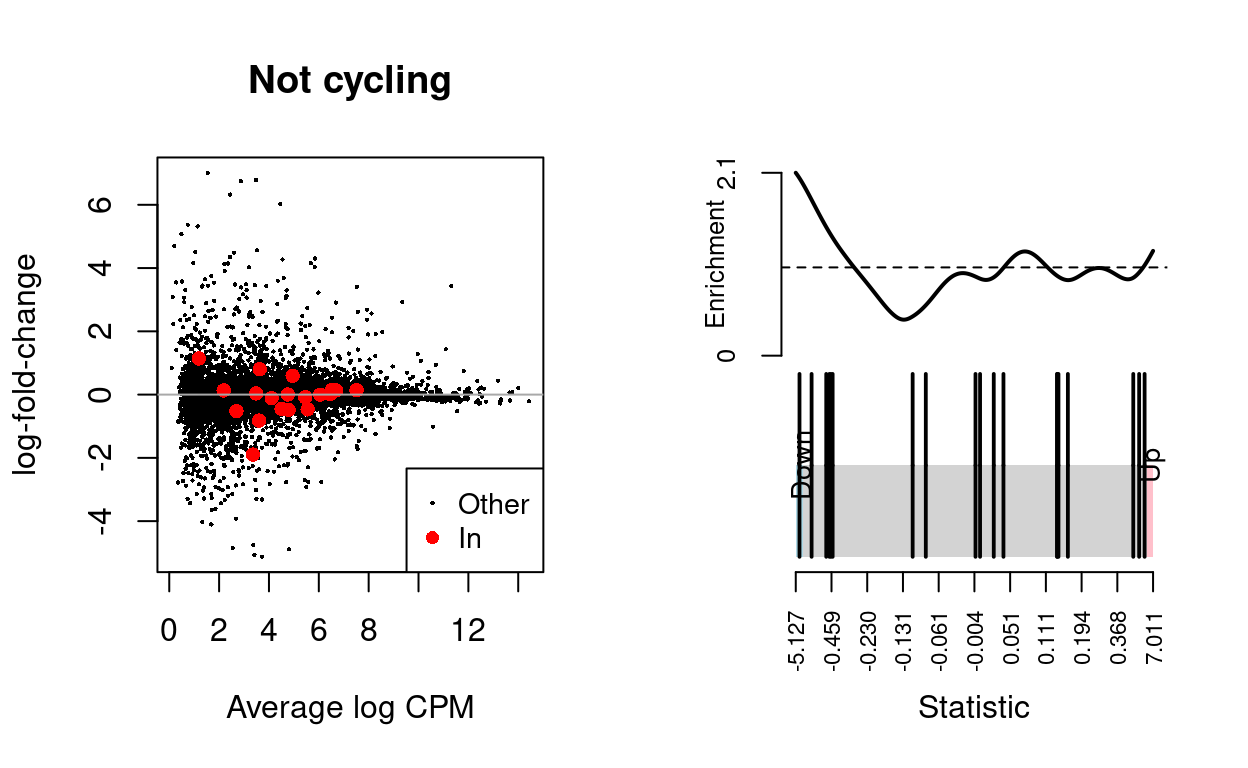
Not cycling
Show code
n <- "Not cycling"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 19 | Down | 0.1278257 | 0.0002985 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
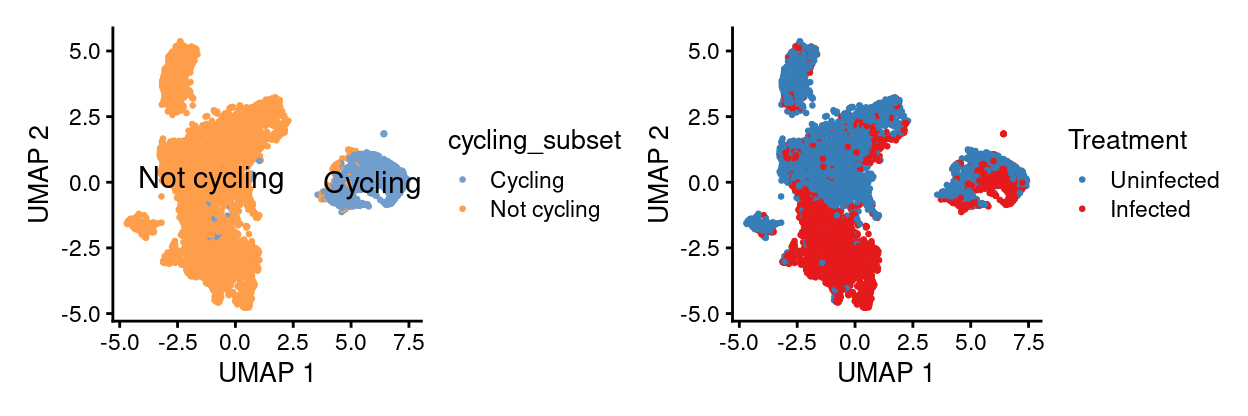
DA
- Number of cells/label/sample
Show code
infected_1 infected_2 infected_3 uninfected_1
Cycling 226 171 327 171
Not cycling 1121 1004 1007 1618
uninfected_2 uninfected_3
Cycling 358 142
Not cycling 1694 1292Show code
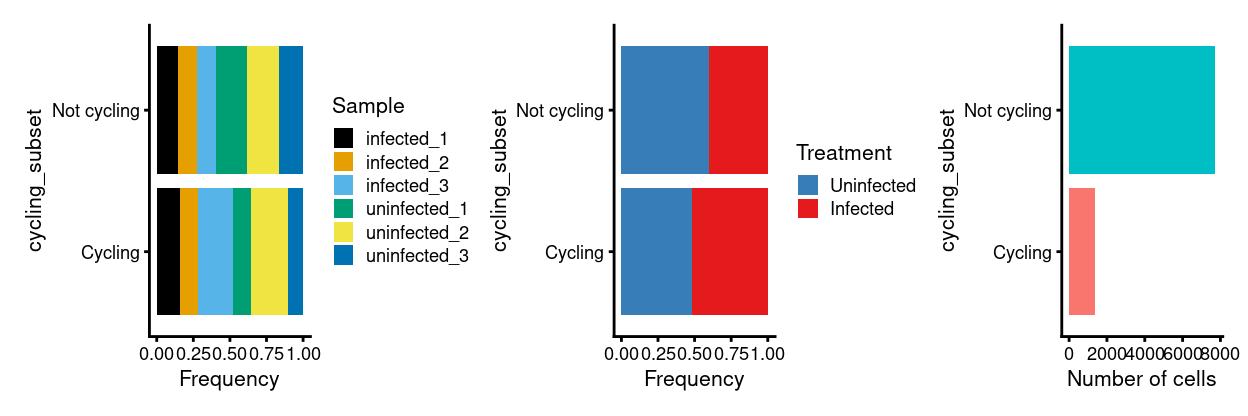
Show code
p3 <- ggplot(as.data.frame(colData(sce)[, c("cycling_subset", "Sample")])) +
geom_bar(
aes(x = cycling_subset, fill = Sample),
position = position_fill(reverse = TRUE)) +
coord_flip() +
ylab("Frequency") +
scale_fill_manual(values = sample_colours) +
theme_cowplot(font_size = 8)
p4 <- ggplot(as.data.frame(colData(sce)[, c("cycling_subset", "Treatment")])) +
geom_bar(
aes(x = cycling_subset, fill = Treatment),
position = position_fill(reverse = TRUE)) +
coord_flip() +
ylab("Frequency") +
scale_fill_manual(values = treatment_colours) +
theme_cowplot(font_size = 8)
p5 <- ggplot(as.data.frame(colData(sce)[, "cycling_subset", drop = FALSE])) +
geom_bar(aes(x = cycling_subset, fill = cycling_subset)) +
coord_flip() +
ylab("Number of cells") +
theme_cowplot(font_size = 8) +
guides(fill = FALSE)
p3 + p4 + p5 + plot_layout(ncol = 3)
TRUE/FALSEif label can be tested for DA
Show code
# NOTE: Can't use quasi-likelihood framework with only two labels.
extra.info <- colData(sce)[match(colnames(abundances), sce$Sample), ]
y.ab <- DGEList(abundances, samples = extra.info)
keep <- filterByExpr(y.ab, group = y.ab$samples$Treatment)
keep
Cycling Not cycling
TRUE TRUE - Number of labels that are DA (
-1means less abundant inInfectedvs.Uninfected,0means not DE,1means more abundant inInfectedvs.Uninfected)
Show code
y.ab <- y.ab[keep,]
design <- model.matrix(~Treatment, y.ab$samples)
y.ab <- estimateDisp(y.ab, design, trend = "none")
fit.ab <- glmFit(y.ab)
res <- glmLRT(fit.ab, coef = "TreatmentInfected")
summary(decideTests(res))
TreatmentInfected
Down 0
NotSig 2
Up 0- Full results (negative
logFCmeans less abundant inInfectedvs.Uninfected, positivelogFCmeans more abundant inInfectedvs.Uninfected)
Show code
topTags(res, n = Inf)
Coefficient: TreatmentInfected
logFC logCPM LR PValue FDR
Cycling 0.5952481 17.24486 2.947518 0.08600953 0.1720191
Not cycling -0.1075603 19.68781 1.001231 0.31701277 0.3170128Analysis based on subcluster labels in Not cycling subset
DE
We require that a gene has an absolute fold change (\(FC\)) \(>1.5\) (i.e. \(|log_2(FC)| > log_2(1.5) \approx 0.58\)) and an \(FDR < 0.05\) to be called as differentially expressed. This is achieved by using the glmTreat() method in edgeR, which is a statistically rigorous method for thresholded differential expression testing.
Please see output/DEGs/not_cycling_subset_subclusters/ for heatmaps and spreadsheets of these DEG lists, including results of GO and KEGG analyses using the goana() and kegga() functions from the limma package. The heatmaps show up to the top-50 DEGs (ordered by \(FDR\)).
This directory also contains interactive Glimma plots of the differential expression results. The Glimma plots show the pseudobulk data for that label.
Show code
# NOTE: Have to drop the complicated TRA and TRB DFrameList objects.
x <- not_cycling_sce
x$TRA <- NULL
x$TRB <- NULL
not_cycling_sce.subcluster_Sample <- aggregateAcrossCells(
x,
id = colData(x)[, c("subcluster", "Sample", "Treatment")],
coldata.merge = FALSE,
use.dimred = FALSE,
use.altexps = FALSE)
sizeFactors(not_cycling_sce.subcluster_Sample) <- NULL
not_cycling_sce.subcluster_Sample <-
logNormCounts(not_cycling_sce.subcluster_Sample)
colLabels(not_cycling_sce.subcluster_Sample) <-
not_cycling_sce.subcluster_Sample$subcluster
colnames(not_cycling_sce.subcluster_Sample) <- paste0(
not_cycling_sce.subcluster_Sample$subcluster,
".",
not_cycling_sce.subcluster_Sample$Sample)
Show code
not_cycling_sce.subcluster_Sample <- runMDS(not_cycling_sce.subcluster_Sample)
p1 <- plotMDS(
not_cycling_sce.subcluster_Sample,
colour_by = "subcluster",
shape_by = "Treatment",
point_size = 3,
point_alpha = 2)
p2 <- plotMDS(
not_cycling_sce.subcluster_Sample,
colour_by = "subcluster",
shape_by = "Sample",
point_size = 3,
point_alpha = 2)
p1 + p2
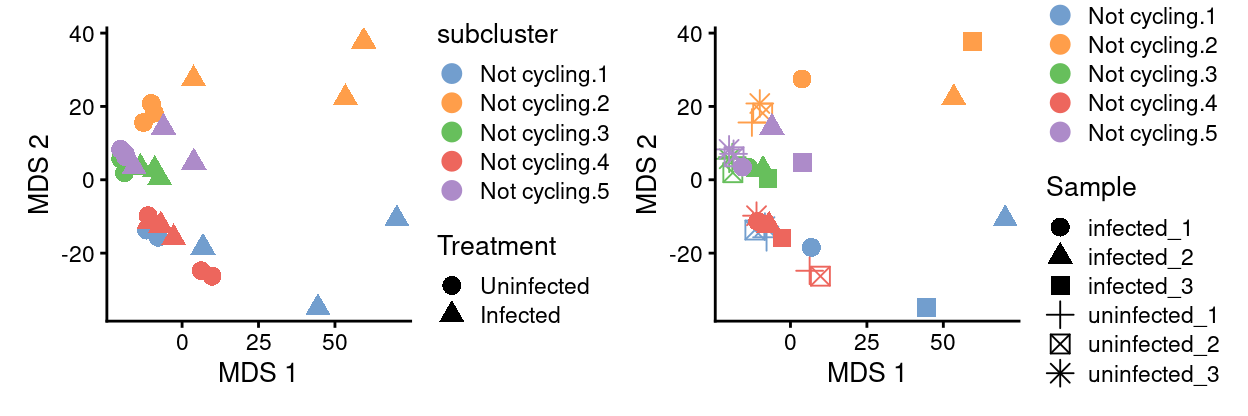
Show code
not_cycling_sce.subcluster_Sample_filt <-
not_cycling_sce.subcluster_Sample[, not_cycling_sce.subcluster_Sample$ncells >= 10]
de_results <- pseudoBulkDGE(
not_cycling_sce.subcluster_Sample_filt,
label = not_cycling_sce.subcluster_Sample_filt$subcluster,
design = ~Treatment,
coef = "TreatmentInfected",
condition = not_cycling_sce.subcluster_Sample_filt$Treatment,
include.intermediates = TRUE,
lfc = log2(1.5))
- Labels for which we couldn’t run DE (
character(0)means none)
Show code
metadata(de_results)$failed
character(0)- Number of DEGs per label (
-1means downregulated inInfectedvs.Uninfected,0means not DE,1means upregulated inInfectedvs.Uninfected)
Show code
is.de <- decideTestsPerLabel(de_results, threshold = 0.05)
summarizeTestsPerLabel(is.de)
-1 0 1 NA
Not cycling.1 1 6120 0 14481
Not cycling.2 2 8254 5 12341
Not cycling.3 10 8829 25 11738
Not cycling.4 3 9086 0 11513
Not cycling.5 3 9465 27 11107Show code
outdir <- here("output", "DEGs", "not_cycling_subset_subcluster")
dir.create(outdir, recursive = TRUE)
createDEGOutputs(
outdir = outdir,
de_results = de_results,
summed_filt = not_cycling_sce.subcluster_Sample_filt,
sce = x,
fdr = 0.05)
Show code
lapply(names(de_results), function(n) {
se <- not_cycling_sce.subcluster_Sample_filt[, not_cycling_sce.subcluster_Sample_filt$label == n]
x <- de_results[[n]]
x <- x[order(x$FDR), ]
features <- rownames(x[which(x$FDR < 0.05), ])
if (length(features) > 2) {
plotHeatmap(
se,
features = head(features, 50),
center = TRUE,
zlim = c(-3, 3),
symmetric = TRUE,
color = hcl.colors(101, "Blue-Red 3"),
order_columns_by = c("Treatment", "Sample"),
column_annotation_colors = list(
Treatment = treatment_colours,
Sample = sample_colours),
cluster_rows = TRUE,
fontsize = 8,
main = n,
filename = file.path(outdir, paste0(n, ".pseudobulk_heatmap.pdf")))
plotGroupedHeatmap(
not_cycling_sce,
features = head(features, 50),
group = "Sample",
columns = not_cycling_sce$subcluster == n,
center = TRUE,
symmetric = TRUE,
color = hcl.colors(101, "Blue-Red 3"),
cluster_rows = TRUE,
fontsize = 8,
main = n,
cluster_cols = FALSE,
filename = file.path(
outdir,
paste0(n, ".average_expression_heatmap.pdf")))
}
})
Gene set testing
We use the fry() function from the edgeR R/Bioconductor package to perform a self-contained gene set test against the null hypothesis that none of the genes in the BCL-family gene set (supplied by James) are differentially expressed. We can visualise the expression of the genes in any given set and in any given comparison by highlighting those genes on the MD plot and/or using a ‘barcode plot.’ Such figures are often included in publications.
Not cycling.1
Show code
n <- "Not cycling.1"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 10 | Up | 0.7725256 | 0.1818233 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
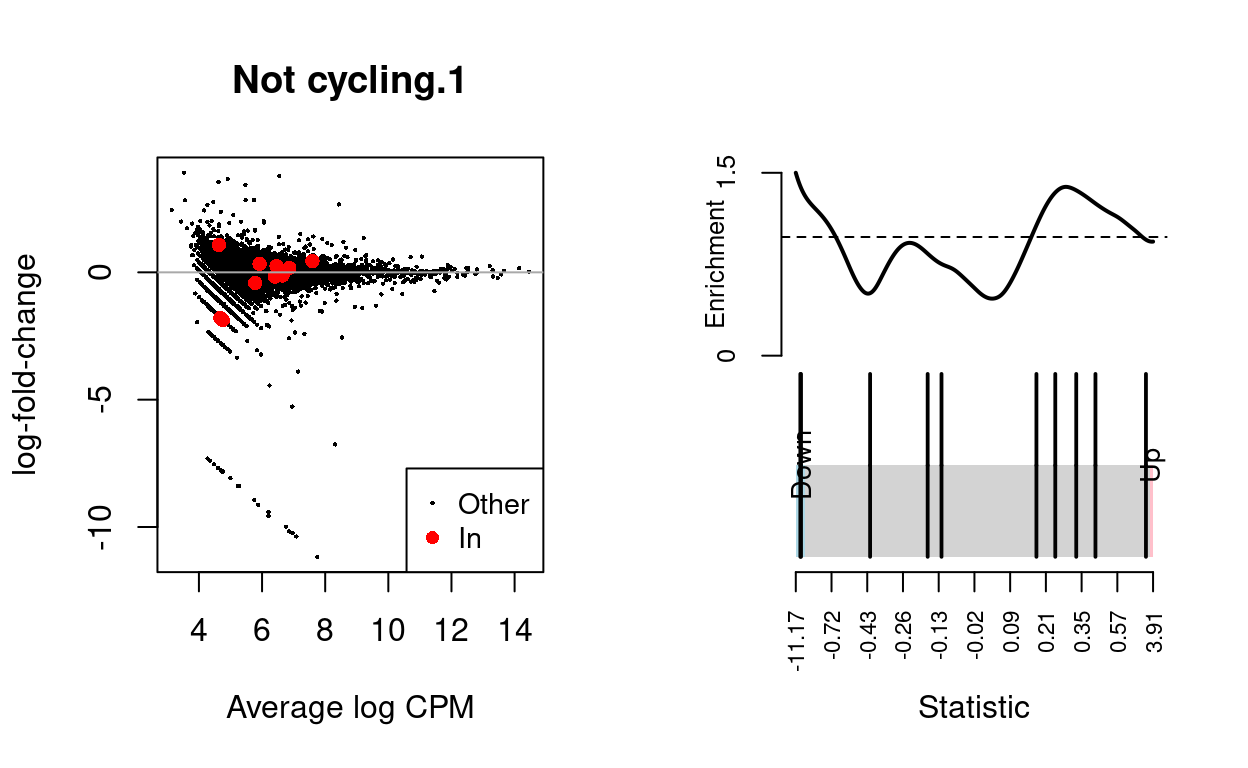
Not cycling.2
Show code
n <- "Not cycling.2"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 16 | Up | 0.5962283 | 0.1791845 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
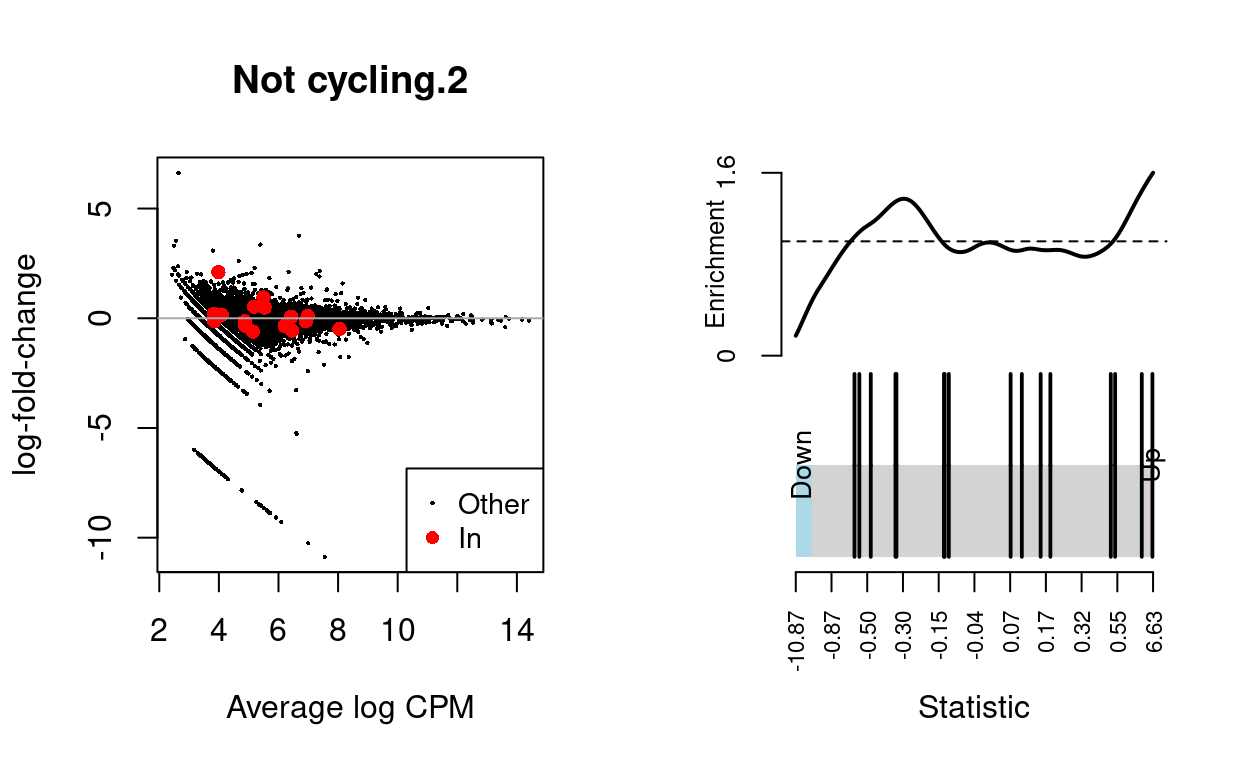
Not cycling.3
Show code
n <- "Not cycling.3"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 16 | Down | 0.5429568 | 0.1337415 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
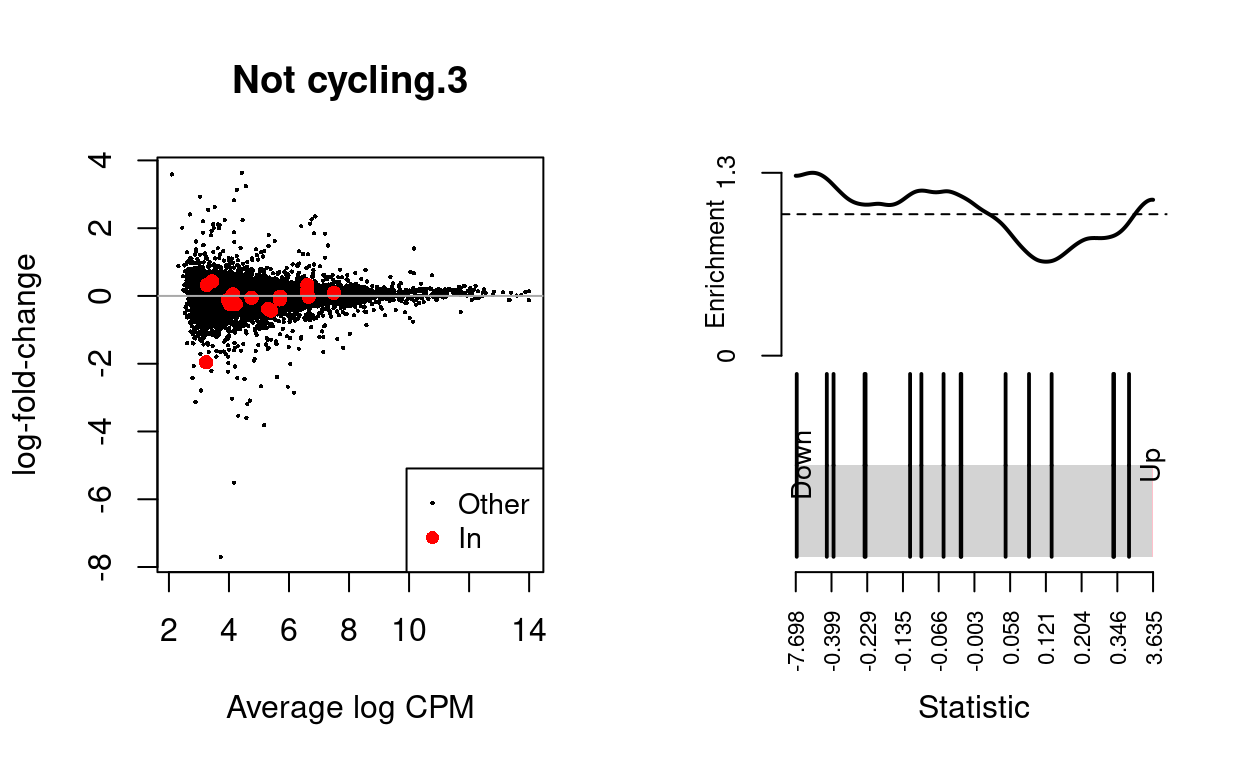
Not cycling.4
Show code
n <- "Not cycling.4"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 17 | Up | 0.5591755 | 0.1480697 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
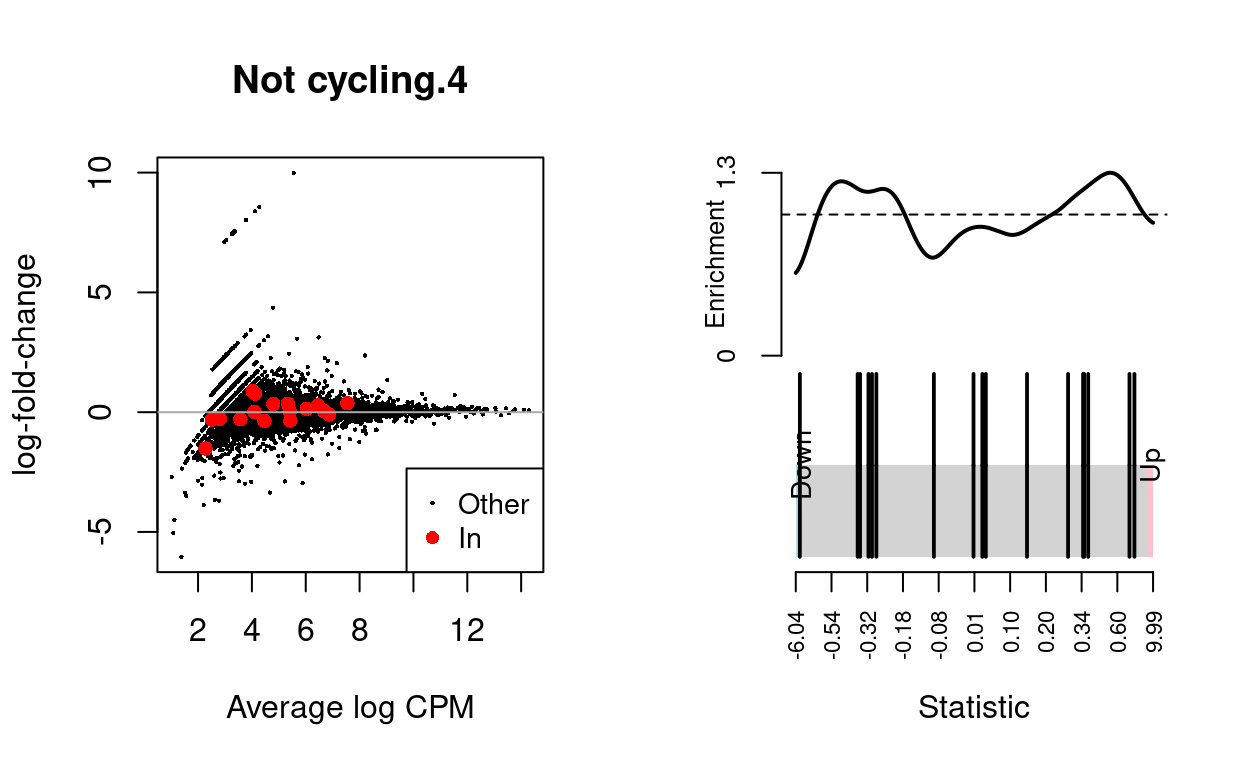
Not cycling.5
Show code
n <- "Not cycling.5"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 17 | Down | 0.2202029 | 0.0323618 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
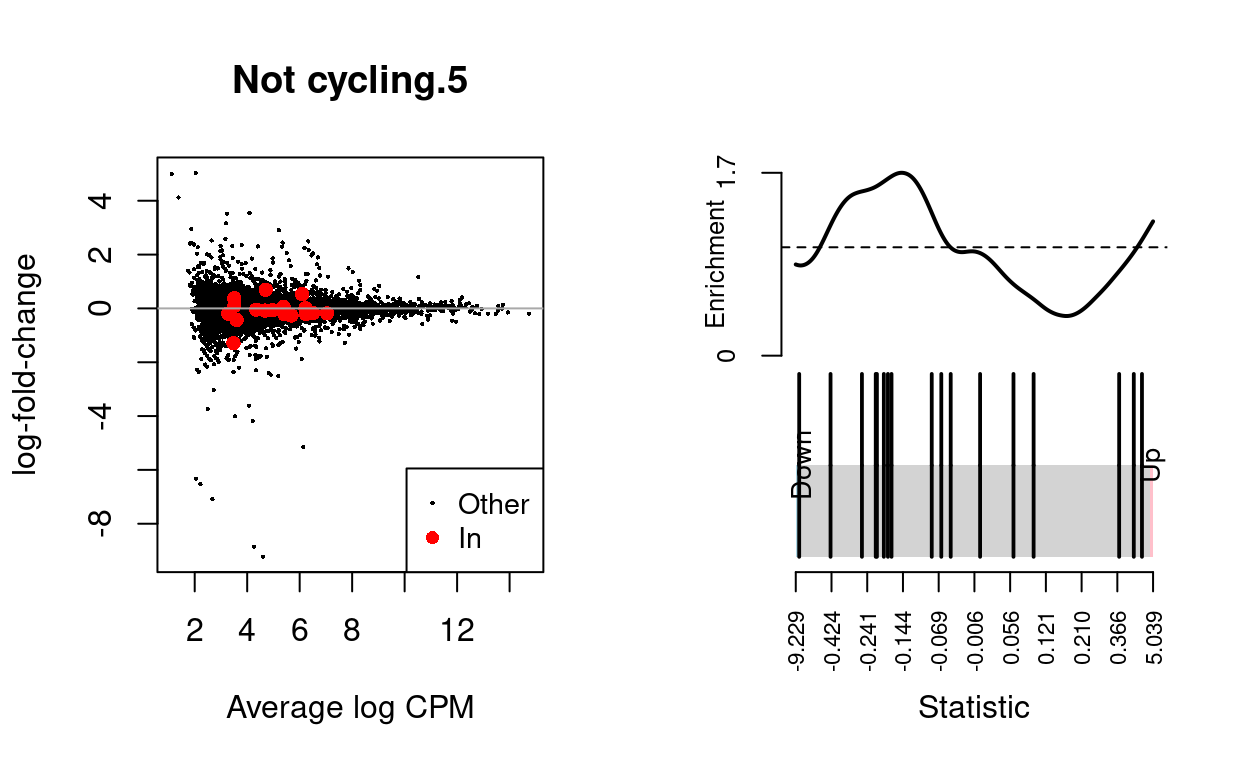
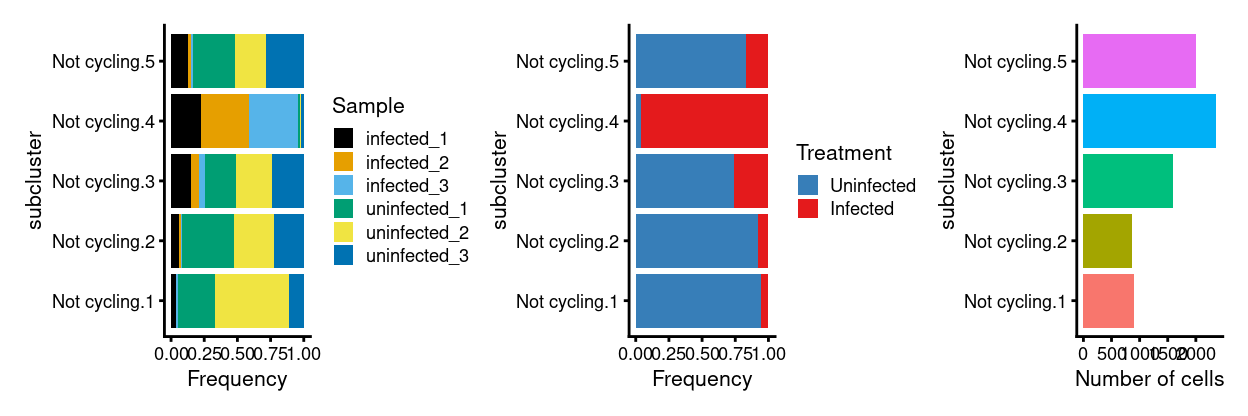
DA
- Number of cells/label/sample
infected_1 infected_2 infected_3 uninfected_1
Not cycling.1 32 3 14 247
Not cycling.2 53 11 7 342
Not cycling.3 241 101 70 366
Not cycling.4 532 848 881 29
Not cycling.5 263 41 35 634
uninfected_2 uninfected_3
Not cycling.1 507 97
Not cycling.2 258 197
Not cycling.3 440 380
Not cycling.4 30 41
Not cycling.5 459 577Show code
Show code
p3 <- ggplot(as.data.frame(colData(x)[, c("subcluster", "Sample")])) +
geom_bar(
aes(x = subcluster, fill = Sample),
position = position_fill(reverse = TRUE)) +
coord_flip() +
ylab("Frequency") +
scale_fill_manual(values = sample_colours) +
theme_cowplot(font_size = 8)
p4 <- ggplot(as.data.frame(colData(x)[, c("subcluster", "Treatment")])) +
geom_bar(
aes(x = subcluster, fill = Treatment),
position = position_fill(reverse = TRUE)) +
coord_flip() +
ylab("Frequency") +
scale_fill_manual(values = treatment_colours) +
theme_cowplot(font_size = 8)
p5 <- ggplot(as.data.frame(colData(x)[, "subcluster", drop = FALSE])) +
geom_bar(aes(x = subcluster, fill = subcluster)) +
coord_flip() +
ylab("Number of cells") +
theme_cowplot(font_size = 8) +
guides(fill = FALSE)
p3 + p4 + p5 + plot_layout(ncol = 3)
TRUE/FALSEif label can be tested for DA
Show code
extra.info <- colData(sce)[match(colnames(abundances), sce$Sample), ]
y.ab <- DGEList(abundances, samples = extra.info)
keep <- filterByExpr(y.ab, group = y.ab$samples$Treatment)
keep
Not cycling.1 Not cycling.2 Not cycling.3 Not cycling.4 Not cycling.5
TRUE TRUE TRUE TRUE TRUE - Number of labels that are DA (
-1means less abundant inInfectedvs.Uninfected,0means not DE,1means more abundant inInfectedvs.Uninfected)
Show code
y.ab <- y.ab[keep,]
design <- model.matrix(~Treatment, y.ab$samples)
y.ab <- estimateDisp(y.ab, design, trend = "none")
fit.ab <- glmFit(y.ab)
res <- glmLRT(fit.ab, coef = "TreatmentInfected")
summary(decideTests(res))
TreatmentInfected
Down 3
NotSig 1
Up 1- Full results (negative
logFCmeans less abundant inInfectedvs.Uninfected, positivelogFCmeans more abundant inInfectedvs.Uninfected)
Show code
topTags(res, n = Inf)
Coefficient: TreatmentInfected
logFC logCPM LR PValue FDR
Not cycling.4 5.023174 18.52370 35.433894 2.638552e-09 1.319276e-08
Not cycling.1 -3.521606 16.56704 19.295114 1.119926e-05 2.799814e-05
Not cycling.2 -2.973163 16.58736 14.604386 1.326055e-04 2.210092e-04
Not cycling.5 -1.835740 17.85842 6.212515 1.268503e-02 1.585629e-02
Not cycling.3 -1.017010 17.57522 2.006615 1.566145e-01 1.566145e-01Analysis based on subcluster labels in Cycling subset
DE
We require that a gene has an absolute fold change (\(FC\)) \(>1.5\) (i.e. \(|log_2(FC)| > log_2(1.5) \approx 0.58\)) and an \(FDR < 0.05\) to be called as differentially expressed. This is achieved by using the glmTreat() method in edgeR, which is a statistically rigorous method for thresholded differential expression testing.
Please see output/DEGs/cycling_subset_subclusters/ for heatmaps and spreadsheets of these DEG lists, including results of GO and KEGG analyses using the goana() and kegga() functions from the limma package. The heatmaps show up to the top-50 DEGs (ordered by \(FDR\)).
This directory also contains interactive Glimma plots of the differential expression results. The Glimma plots show the pseudobulk data for that label.
Show code
# NOTE: Have to drop the complicated TRA and TRB DFrameList objects.
x <- cycling_sce
x$TRA <- NULL
x$TRB <- NULL
cycling_sce.subcluster_Sample <- aggregateAcrossCells(
x,
id = colData(x)[, c("subcluster", "Sample", "Treatment")],
coldata.merge = FALSE,
use.dimred = FALSE,
use.altexps = FALSE)
sizeFactors(cycling_sce.subcluster_Sample) <- NULL
cycling_sce.subcluster_Sample <- logNormCounts(cycling_sce.subcluster_Sample)
colLabels(cycling_sce.subcluster_Sample) <-
cycling_sce.subcluster_Sample$subcluster
colnames(cycling_sce.subcluster_Sample) <- paste0(
cycling_sce.subcluster_Sample$subcluster,
".",
cycling_sce.subcluster_Sample$Sample)
Show code
cycling_sce.subcluster_Sample <- runMDS(cycling_sce.subcluster_Sample)
p1 <- plotMDS(
cycling_sce.subcluster_Sample,
colour_by = "subcluster",
shape_by = "Treatment",
point_size = 3,
point_alpha = 2)
p2 <- plotMDS(
cycling_sce.subcluster_Sample,
colour_by = "subcluster",
shape_by = "Sample",
point_size = 3,
point_alpha = 2)
p1 + p2
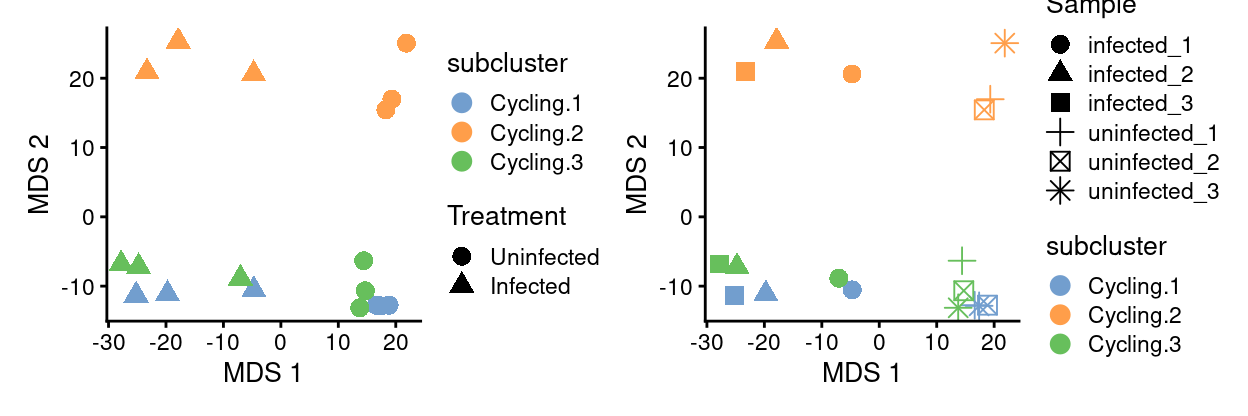
Show code
cycling_sce.subcluster_Sample_filt <-
cycling_sce.subcluster_Sample[, cycling_sce.subcluster_Sample$ncells >= 10]
de_results <- pseudoBulkDGE(
cycling_sce.subcluster_Sample_filt,
label = cycling_sce.subcluster_Sample_filt$subcluster,
design = ~Treatment,
coef = "TreatmentInfected",
condition = cycling_sce.subcluster_Sample_filt$Treatment,
include.intermediates = TRUE,
lfc = log2(1.5))
- Labels for which we couldn’t run DE (
character(0)means none)
Show code
metadata(de_results)$failed
character(0)- Number of DEGs per label (
-1means downregulated inInfectedvs.Uninfected,0means not DE,1means upregulated inInfectedvs.Uninfected)
Show code
is.de <- decideTestsPerLabel(de_results, threshold = 0.05)
summarizeTestsPerLabel(is.de)
-1 0 1 NA
Cycling.1 27 7659 29 12887
Cycling.2 11 6184 20 14387
Cycling.3 14 8500 29 12059Show code
outdir <- here("output", "DEGs", "cycling_subset_subcluster")
dir.create(outdir, recursive = TRUE)
createDEGOutputs(
outdir = outdir,
de_results = de_results,
summed_filt = cycling_sce.subcluster_Sample_filt,
sce = x,
fdr = 0.05)
Show code
lapply(names(de_results), function(n) {
se <- cycling_sce.subcluster_Sample_filt[, cycling_sce.subcluster_Sample_filt$label == n]
x <- de_results[[n]]
x <- x[order(x$FDR), ]
features <- rownames(x[which(x$FDR < 0.05), ])
if (length(features) > 2) {
plotHeatmap(
se,
features = head(features, 50),
center = TRUE,
zlim = c(-3, 3),
symmetric = TRUE,
color = hcl.colors(101, "Blue-Red 3"),
order_columns_by = c("Treatment", "Sample"),
column_annotation_colors = list(
Treatment = treatment_colours,
Sample = sample_colours),
cluster_rows = TRUE,
fontsize = 8,
main = n,
filename = file.path(outdir, paste0(n, ".pseudobulk_heatmap.pdf")))
plotGroupedHeatmap(
cycling_sce,
features = head(features, 50),
group = "Sample",
columns = cycling_sce$subcluster == n,
center = TRUE,
symmetric = TRUE,
color = hcl.colors(101, "Blue-Red 3"),
cluster_rows = TRUE,
fontsize = 8,
main = n,
cluster_cols = FALSE,
filename = file.path(
outdir,
paste0(n, ".average_expression_heatmap.pdf")))
}
})
Gene set testing
We use the fry() function from the edgeR R/Bioconductor package to perform a self-contained gene set test against the null hypothesis that none of the genes in the BCL-family gene set (supplied by James) are differentially expressed. We can visualise the expression of the genes in any given set and in any given comparison by highlighting those genes on the MD plot and/or using a ‘barcode plot.’ Such figures are often included in publications.
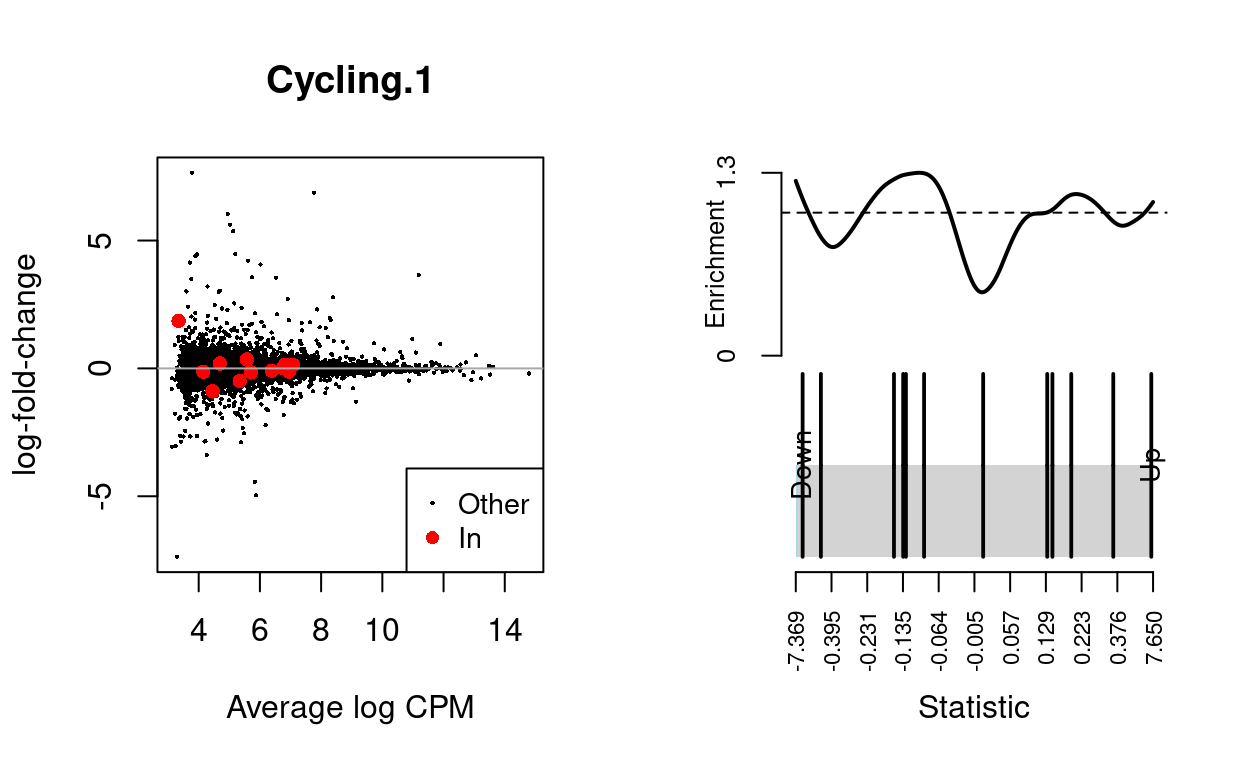
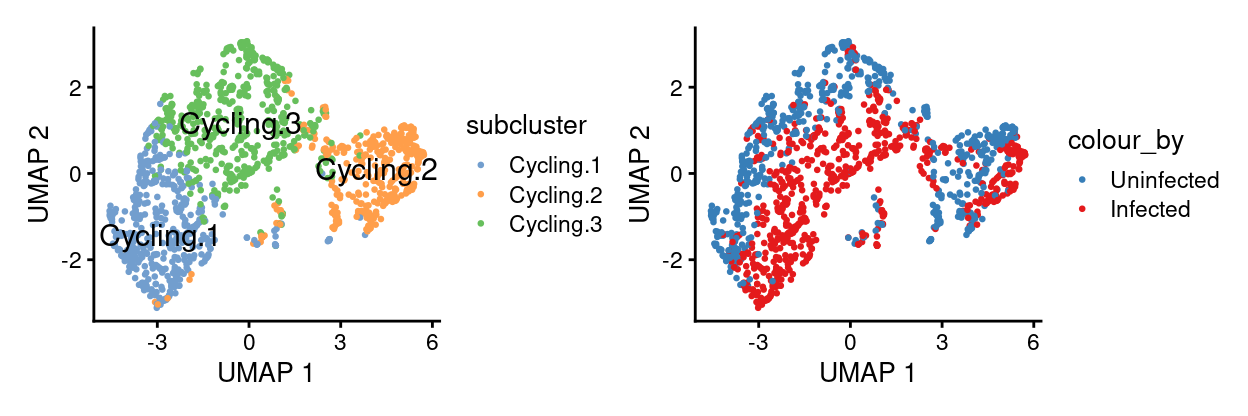
Cycling.1
Show code
n <- "Cycling.1"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 12 | Up | 0.7233728 | 0.0026638 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
Cycling.2
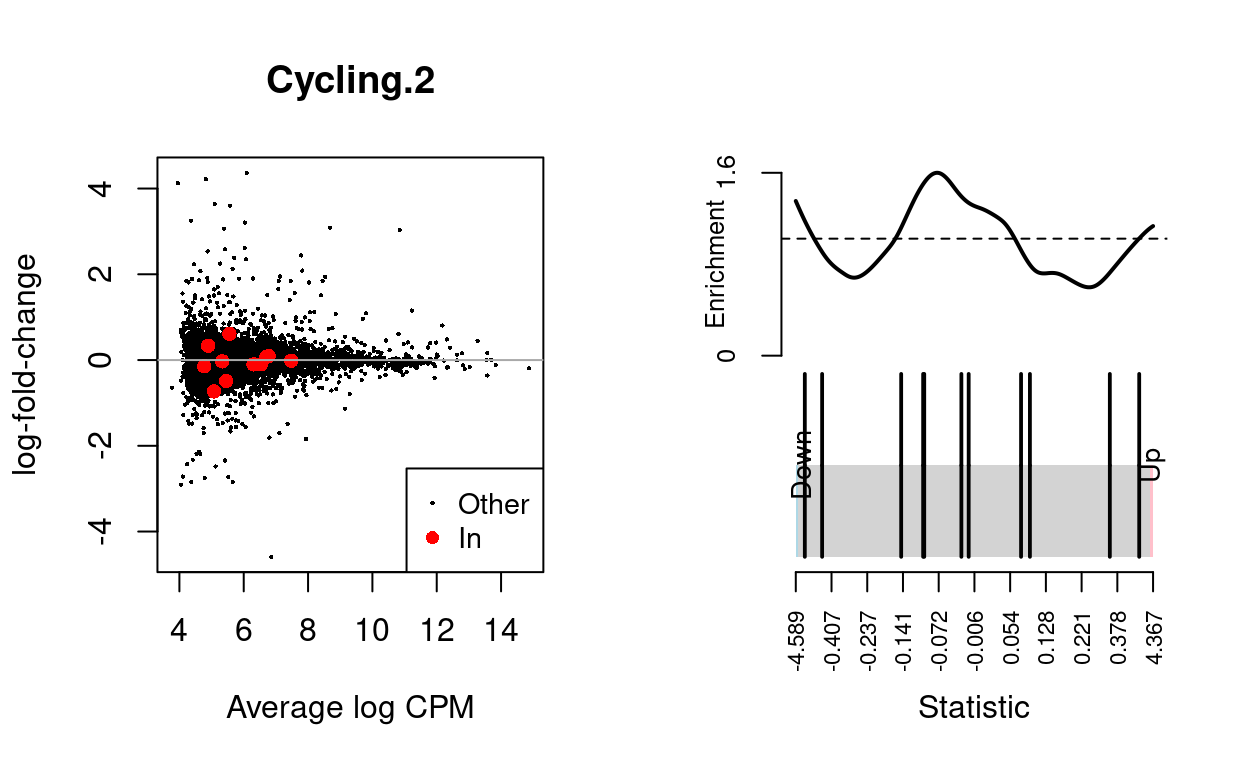
Show code
n <- "Cycling.2"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 11 | Down | 0.7174617 | 0.062013 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
Cycling.3
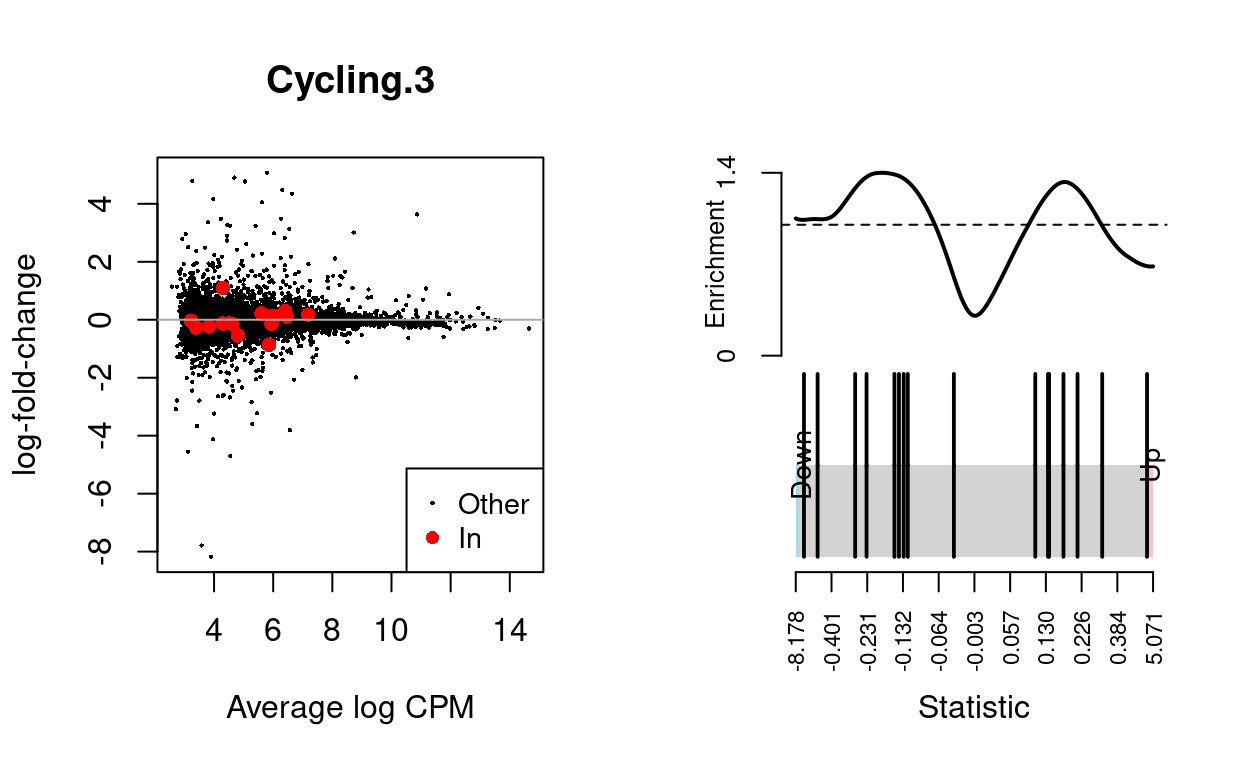
Show code
n <- "Cycling.3"
md <- metadata(de_results[[n]])
y <- md$y
y_design <- md$design
y_index <- ids2indices(
list(`BCL2-family` = bcl2_df$Approved.symbol),
rownames(y))
fry(
y,
index = y_index,
design = y_design,
coef = "TreatmentInfected") %>%
knitr::kable(
caption = "Results of applying `fry()` to the RNA-seq differential expression results using the supplied gene sets. A significant 'Up' P-value means that the differential expression results found in the RNA-seq data are positively correlated with the expression signature from the corresponding gene set. Conversely, a significant 'Down' P-value means that the differential expression log-fold-changes are negatively correlated with the expression signature from the corresponding gene set. A significant 'Mixed' P-value means that the genes in the set tend to be differentially expressed without regard for direction.")
| NGenes | Direction | PValue | PValue.Mixed | |
|---|---|---|---|---|
| BCL2-family | 16 | Down | 0.8767046 | 0.102535 |
Show code
dgelrt <- glmTreat(md$fit, coef = "TreatmentInfected", lfc = log2(1.5))
par(mfrow = c(1, 2))
y_status <- rep("Other", nrow(dgelrt))
y_status[rownames(dgelrt) %in% bcl2_df$Approved.symbol] <- "In"
plotMD(
dgelrt,
status = y_status,
values = "In",
hl.col = "red",
legend = "bottomright",
main = n)
abline(h = 0, col = "darkgrey")
barcodeplot(
statistics = dgelrt$table$logFC,
index = rownames(dgelrt) %in% bcl2_df$Approved.symbol)
DA
- Number of cells/label/sample
infected_1 infected_2 infected_3 uninfected_1
Cycling.1 73 65 147 42
Cycling.2 72 45 55 55
Cycling.3 81 61 125 74
uninfected_2 uninfected_3
Cycling.1 112 48
Cycling.2 113 43
Cycling.3 133 51Show code
Show code
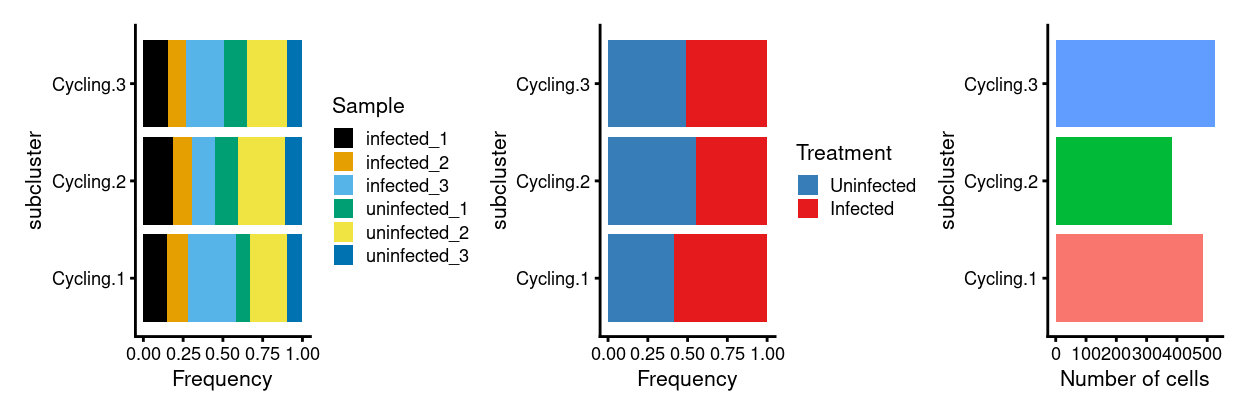
p3 <- ggplot(as.data.frame(colData(x)[, c("subcluster", "Sample")])) +
geom_bar(
aes(x = subcluster, fill = Sample),
position = position_fill(reverse = TRUE)) +
coord_flip() +
ylab("Frequency") +
scale_fill_manual(values = sample_colours) +
theme_cowplot(font_size = 8)
p4 <- ggplot(as.data.frame(colData(x)[, c("subcluster", "Treatment")])) +
geom_bar(
aes(x = subcluster, fill = Treatment),
position = position_fill(reverse = TRUE)) +
coord_flip() +
ylab("Frequency") +
scale_fill_manual(values = treatment_colours) +
theme_cowplot(font_size = 8)
p5 <- ggplot(as.data.frame(colData(x)[, "subcluster", drop = FALSE])) +
geom_bar(aes(x = subcluster, fill = subcluster)) +
coord_flip() +
ylab("Number of cells") +
theme_cowplot(font_size = 8) +
guides(fill = FALSE)
p3 + p4 + p5 + plot_layout(ncol = 3)
TRUE/FALSEif label can be tested for DA
Show code
extra.info <- colData(sce)[match(colnames(abundances), sce$Sample), ]
y.ab <- DGEList(abundances, samples = extra.info)
keep <- filterByExpr(y.ab, group = y.ab$samples$Treatment)
keep
Cycling.1 Cycling.2 Cycling.3
TRUE TRUE TRUE - Number of labels that are DA (
-1means less abundant inInfectedvs.Uninfected,0means not DE,1means more abundant inInfectedvs.Uninfected)
Show code
y.ab <- y.ab[keep,]
design <- model.matrix(~Treatment, y.ab$samples)
y.ab <- estimateDisp(y.ab, design, trend = "none")
fit.ab <- glmFit(y.ab)
res <- glmLRT(fit.ab, coef = "TreatmentInfected")
summary(decideTests(res))
TreatmentInfected
Down 0
NotSig 3
Up 0- Full results (negative
logFCmeans less abundant inInfectedvs.Uninfected, positivelogFCmeans more abundant inInfectedvs.Uninfected)
Show code
topTags(res, n = Inf)
Coefficient: TreatmentInfected
logFC logCPM LR PValue FDR
Cycling.1 0.37294181 18.40769 3.7213695 0.05372029 0.1611609
Cycling.2 -0.35382545 18.10346 2.1597911 0.14166395 0.2124959
Cycling.3 -0.06526804 18.53074 0.2197015 0.63926749 0.6392675Concluding remarks
Show code
saveRDS(
sce.ignoring_cycling_subset,
here("data", "SCEs", "C057_Cooney.ignoring_cycling_subset.sce.rds"),
compress = "xz")
saveRDS(
sce.cycling_subset_Sample,
here("data", "SCEs", "C057_Cooney.cycling_subset_Sample.sce.rds"),
compress = "xz")
saveRDS(
not_cycling_sce.subcluster_Sample,
here("data", "SCEs", "C057_Cooney.not_cycling.subcluster_Sample.sce.rds"),
compress = "xz")
saveRDS(
cycling_sce.subcluster_Sample,
here("data", "SCEs", "C057_Cooney.cycling.subcluster_Sample.sce.rds"),
compress = "xz")
The aggregated SummarizedExperiment objects are available (see data/SCEs/C057_Cooney.ignoring_cycling_subset.sce.rds, data/SCEs/C057_Cooney.cycling_subset_Sample.sce.rds, data/SCEs/C057_Cooney.not_cycling.subcluster_Sample.sce.rds, and data/SCEs/C057_Cooney.cycling.subcluster_Sample.sce.rds). These can be used for visualizations with iSEE.
The output/DEGs/ directory contains CSV files summarising the differential expression results and PDFs showing the pseudobulk expression data for each DEG.
Additional information
The following are available on request:
- Full CSV tables of any data presented.
- PDF/PNG files of any static plots.
Session info
Show code
sessioninfo::session_info()
─ Session info ─────────────────────────────────────────────────────
setting value
version R version 4.0.3 (2020-10-10)
os CentOS Linux 7 (Core)
system x86_64, linux-gnu
ui X11
language (EN)
collate en_US.UTF-8
ctype en_US.UTF-8
tz Australia/Melbourne
date 2021-12-03
─ Packages ─────────────────────────────────────────────────────────
! package * version date lib
P annotate 1.68.0 2020-10-27 [?]
P AnnotationDbi 1.52.0 2020-10-27 [?]
P assertthat 0.2.1 2019-03-21 [?]
P beachmat 2.6.4 2020-12-20 [?]
P beeswarm 0.2.3 2016-04-25 [?]
P Biobase * 2.50.0 2020-10-27 [?]
BiocGenerics * 0.36.0 2020-10-27 [1]
P BiocManager 1.30.10 2019-11-16 [?]
P BiocNeighbors 1.8.2 2020-12-07 [?]
BiocParallel 1.24.1 2020-11-06 [1]
P BiocSingular 1.6.0 2020-10-27 [?]
BiocStyle 2.18.1 2020-11-24 [1]
P bit 4.0.4 2020-08-04 [?]
P bit64 4.0.5 2020-08-30 [?]
P bitops 1.0-6 2013-08-17 [?]
P blob 1.2.1 2020-01-20 [?]
P bluster 1.0.0 2020-10-27 [?]
P bslib 0.2.4 2021-01-25 [?]
P cachem 1.0.4 2021-02-13 [?]
P cli 2.3.1 2021-02-23 [?]
P colorspace 2.0-0 2020-11-11 [?]
P cowplot * 1.1.1 2020-12-30 [?]
P crayon 1.4.1 2021-02-08 [?]
P DBI 1.1.1 2021-01-15 [?]
P DelayedArray 0.16.2 2021-02-26 [?]
P DelayedMatrixStats 1.12.3 2021-02-03 [?]
P DESeq2 1.30.1 2021-02-19 [?]
P digest 0.6.27 2020-10-24 [?]
P distill 1.2 2021-01-13 [?]
P downlit 0.2.1 2020-11-04 [?]
P dplyr 1.0.4 2021-02-02 [?]
P dqrng 0.2.1 2019-05-17 [?]
P edgeR * 3.32.1 2021-01-14 [?]
P ellipsis 0.3.1 2020-05-15 [?]
P evaluate 0.14 2019-05-28 [?]
P fansi 0.4.2 2021-01-15 [?]
P farver 2.1.0 2021-02-28 [?]
P fastmap 1.1.0 2021-01-25 [?]
P genefilter 1.72.1 2021-01-21 [?]
P geneplotter 1.68.0 2020-10-27 [?]
P generics 0.1.0 2020-10-31 [?]
P GenomeInfoDb * 1.26.2 2020-12-08 [?]
GenomeInfoDbData 1.2.4 2021-02-04 [1]
P GenomicRanges * 1.42.0 2020-10-27 [?]
P ggbeeswarm 0.6.0 2017-08-07 [?]
P ggplot2 * 3.3.3 2020-12-30 [?]
P Glimma 2.0.0 2020-10-27 [?]
P glue 1.4.2 2020-08-27 [?]
P gridExtra 2.3 2017-09-09 [?]
P gtable 0.3.0 2019-03-25 [?]
P here * 1.0.1 2020-12-13 [?]
P highr 0.8 2019-03-20 [?]
P htmltools 0.5.1.1 2021-01-22 [?]
P htmlwidgets 1.5.3 2020-12-10 [?]
P httr 1.4.2 2020-07-20 [?]
P igraph 1.2.6 2020-10-06 [?]
P IRanges * 2.24.1 2020-12-12 [?]
P irlba 2.3.3 2019-02-05 [?]
P jquerylib 0.1.3 2020-12-17 [?]
P jsonlite 1.7.2 2020-12-09 [?]
P knitr 1.31 2021-01-27 [?]
P labeling 0.4.2 2020-10-20 [?]
P lattice 0.20-41 2020-04-02 [?]
P lifecycle 1.0.0 2021-02-15 [?]
limma * 3.46.0 2020-10-27 [1]
P locfit 1.5-9.4 2020-03-25 [?]
P magrittr * 2.0.1 2020-11-17 [?]
P Matrix 1.3-2 2021-01-06 [?]
MatrixGenerics * 1.2.1 2021-01-30 [1]
P matrixStats * 0.58.0 2021-01-29 [?]
P memoise 2.0.0 2021-01-26 [?]
P munsell 0.5.0 2018-06-12 [?]
P patchwork * 1.1.1 2020-12-17 [?]
P pheatmap 1.0.12 2019-01-04 [?]
P pillar 1.5.0 2021-02-22 [?]
P pkgconfig 2.0.3 2019-09-22 [?]
P purrr 0.3.4 2020-04-17 [?]
P R6 2.5.0 2020-10-28 [?]
P RColorBrewer 1.1-2 2014-12-07 [?]
P Rcpp 1.0.6 2021-01-15 [?]
P RCurl 1.98-1.2 2020-04-18 [?]
P rlang 0.4.10 2020-12-30 [?]
P rmarkdown 2.11 2021-09-14 [?]
P rprojroot 2.0.2 2020-11-15 [?]
P RSQLite 2.2.3 2021-01-24 [?]
P rsvd 1.0.3 2020-02-17 [?]
P S4Vectors * 0.28.1 2020-12-09 [?]
P sass 0.3.1 2021-01-24 [?]
P scales 1.1.1 2020-05-11 [?]
P scater * 1.18.6 2021-02-26 [?]
P scran * 1.18.5 2021-02-04 [?]
P scuttle 1.0.4 2020-12-17 [?]
P sessioninfo 1.1.1 2018-11-05 [?]
P SingleCellExperiment * 1.12.0 2020-10-27 [?]
P sparseMatrixStats 1.2.1 2021-02-02 [?]
P statmod 1.4.35 2020-10-19 [?]
P stringi 1.5.3 2020-09-09 [?]
P stringr 1.4.0 2019-02-10 [?]
P SummarizedExperiment * 1.20.0 2020-10-27 [?]
P survival 3.2-7 2020-09-28 [?]
P tibble 3.1.0 2021-02-25 [?]
P tidyselect 1.1.0 2020-05-11 [?]
P utf8 1.1.4 2018-05-24 [?]
P vctrs 0.3.6 2020-12-17 [?]
P vipor 0.4.5 2017-03-22 [?]
P viridis 0.5.1 2018-03-29 [?]
P viridisLite 0.3.0 2018-02-01 [?]
P withr 2.4.1 2021-01-26 [?]
P xaringanExtra 0.5.5 2021-12-03 [?]
P xfun 0.28 2021-11-04 [?]
P XML 3.99-0.5 2020-07-23 [?]
P xtable 1.8-4 2019-04-21 [?]
P XVector 0.30.0 2020-10-27 [?]
P yaml 2.2.1 2020-02-01 [?]
P zlibbioc 1.36.0 2020-10-27 [?]
source
Bioconductor
Bioconductor
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
Bioconductor
Bioconductor
CRAN (R 4.0.0)
Bioconductor
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.2)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
Bioconductor
CRAN (R 4.0.3)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.2)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.2)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.3)
CRAN (R 4.0.0)
Bioconductor
Bioconductor
Bioconductor
CRAN (R 4.0.0)
Bioconductor
Bioconductor
CRAN (R 4.0.2)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.2)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
CRAN (R 4.0.3)
Github (gadenbuie/xaringanExtra@cc1f613)
CRAN (R 4.0.3)
CRAN (R 4.0.0)
CRAN (R 4.0.0)
Bioconductor
CRAN (R 4.0.0)
Bioconductor
[1] /stornext/Projects/score/Analyses/C057_Cooney/renv/library/R-4.0/x86_64-pc-linux-gnu
[2] /tmp/RtmpyCwH1Q/renv-system-library
[3] /stornext/System/data/apps/R/R-4.0.3/lib64/R/library
P ── Loaded and on-disk path mismatch.